

ADSL Cure Odyssey
A scientist and mother from Belgrade reached out last year about therapeutic options for her daughter suffering from an ultra-rare disease called ADSL deficiency. Last week, we finally got yeast hits.

In collaboration with


 / Twitter")

Disclaimer
The results of the ADSL deficiency drug repurposing project that we are sharing in the spirit of open science below are novel preclinical research findings and therefore they do not constitute the practice of medicine. Please consult your physician or clinical care team if you’re considering off-label use of any approved drug. The same caution applies to nutraceuticals, supplements and “generally recognized as safe” compounds.
We received an email inquiry almost one year ago from Iva Milacic asking for help. Her daughter Minja has ADSL deficiency, an inborn error of purine metabolism. Mutations in ADSL are associated with severe intellectual disability (ID), epilepsy, ataxia and autistic features. There’s a wide spectrum of severity and time of onset. Like all the other diseases we work on, there are no approved treatments. That means the clock is ticking.
The A’s and G’s of the 4-letter DNA code are purines, chemically speaking. Purines are required for DNA replication, which is of course a requirement for life. Purines have other roles in cellular metabolism. For example, we all remember from high school biology that the energy currency of the cell is ATP, which is made from AMP, one of the products of ADSL catalysis. Therefore, like most other inborn errors of metabolism, patients with ADSL deficiency must have at least some residual enzymatic activity.
The ADSL gene encodes a protein called adenylosuccinate lyase. This enzyme is responsible for two non-sequential steps in the purine biosynthesis pathway: the conversion of succinylaminoimidazole carboxamide ribotide (SAICAR) to aminoimidazole carboxamide ribotide (AICAR), and the conversion of adenylosuccinate (S-AMP) to adenosine monophosphate (AMP).

ADSL deficiency may in fact be a chameleon: multiple metabolic imbalances that manifest in some tissues or cell types or times during life but not others. On the upper branch of purine metabolism, a toxic metabolic called succinylaminoimidazole carboxamide riboside (SAICAr) accumulates when ADSL activity is diminished by mutation. On the lower branch of purine metabolism, ADSL deficiency leads to a buildup of succinyladenosine (S-Ado). Levels of the key metabolite fumarate are also reduced by ADSL deficiency. The ripple effects likely extend beyond just the purine cycle.
One thing goes up. Another thing goes down. Posed in more technical terms: is the primary disease driver of ADSL deficiency the accumulation of toxic metabolites or the depletion of essential metabolic end-products like nucleosides or fumarate?
Or do metabolic imbalances in both directions — too much of something unwanted and too little of something desirable — act as co-conspirators? Although measurements of purine levels in some tissues from patients with ADSL deficiency suggest that compensatory or bypass pathways kick in to ensure purine levels remain steady, the jury is still out.
All that to say we hadn’t ever worked on the purine biosynthetic pathway, which initially gave us pause. But we did have lots of experience with other inborn errors of metabolism. On paper, ADSL deficiency seemed to be a great fit for modeling in a yeast patient avatar. ADSL is conserved across the animal kingdom and particularly in yeast such that, the human ADSL gene can complement yeast ADE13 function.
The genome of budding yeast (Saccharomyces cerevisiae) contains an ADSL ortholog called ADE13 in which the corresponding mutation would be introduced based on the protein sequence alignment below.

In the paternally inherited mutation Y114H, the tyrosine (Y) residue at position 114 in the human ADSL gene corresponds to the phenylalanine (F) at position 111 in the yeast ADE13 gene. The Y-to-F substitution is a conserved substitution. The amino acid though different is structurally and functionally alike and occurs in similar protein folds. The amino acid histidine (H) cannot functionally substitute or be substituted by any other amino acid and is hence likely to impair ADE13 function. More straightforwardly, the maternally inherited mutation L121F is conserved in yeast and other species, and corresponds to L118 in ADE13.
We initially thought that yeast engineered to express the patient’s Y144H and L121F mutations would be the fastest turnaround and most cost-effective disease models in which to characterize the mutations. Turns out we were mistaken. Trudging through the trough of troubleshooting for most of this year, there were times when it felt like we’d exhausted all available options. But we persevered. We had no choice. We couldn’t let Minja down.

Based on our contemporaneous experiences with PGAP3-CDG, PIGA-CDG, PIGN-CDG and PIGS-CDG, we knew that the DAmP mutant collection was a way to assess a generic hypomorphic variant. We also learned that sensitization was a way to ensure a screenotype robust enough for drug screening. Recall that the DAmP collections contains genomic disruptions in essential yeast genes that result in ~10% residual expression and a growth defect.
Assay development was initially smooth sailing back in February. We predicted that the ADE13 DAmP mutant would grow more poorly when deprived of dietary adenine and when exposed to high temperature (37˚C). Our intuition was correct. Adenine withdrawal from the media gives a boost to wildtype (BY4741) relative to the ADE13 DAmP mutant strains. The result was a nearly 10-fold assay window. Behind schedule at this point, we thought the worst was behind us.
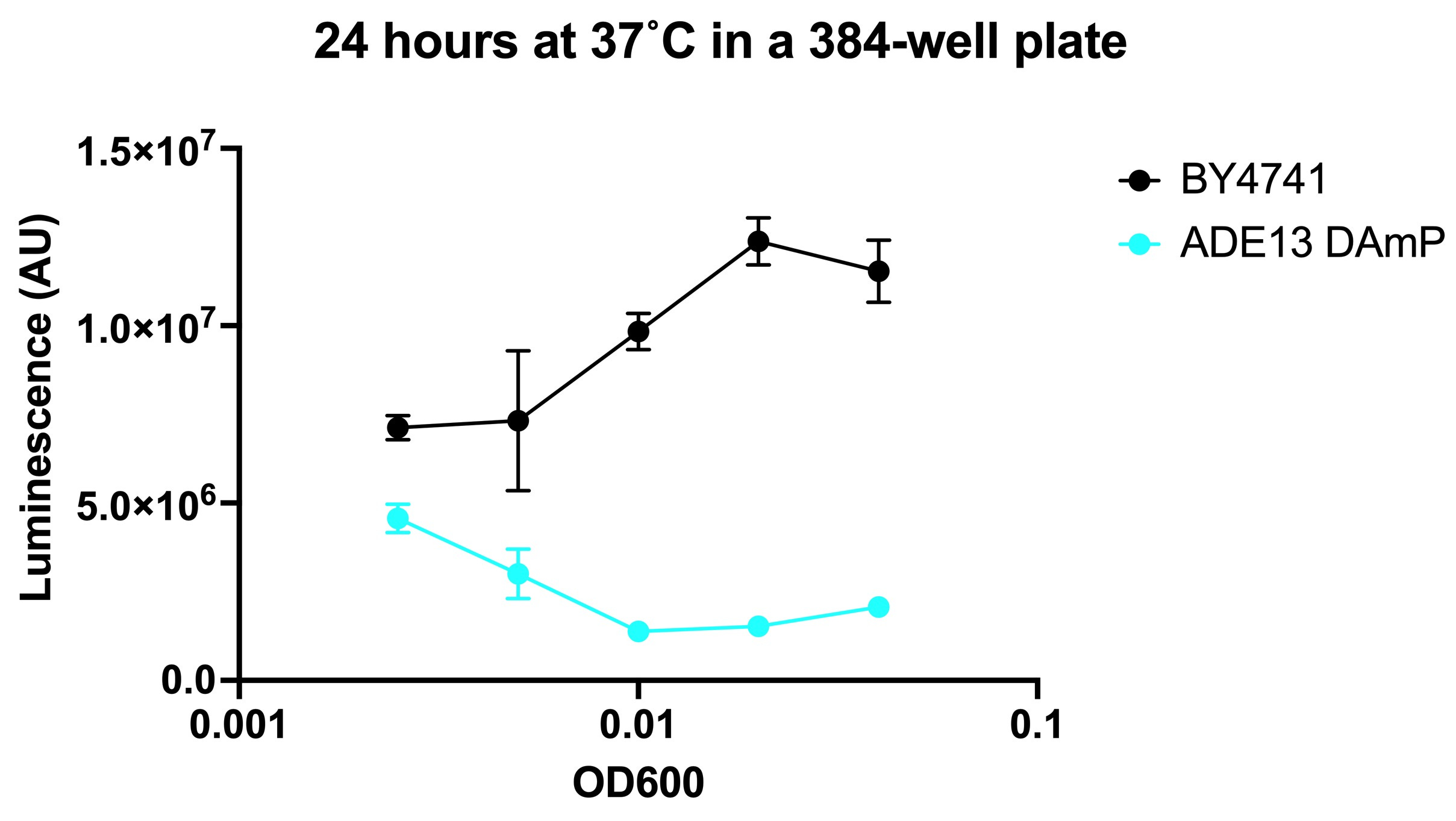
Here are the results of the seeding density optimization from April. This was supposed to be the final step before drug screening. Experiments were performed by Dr Jez Revalde at the SMDC at UCSF.

We ran headfirst into a paradoxical result. The ADE13 DAmP mutant grew more slowly than wildtype over a short incubation (several hours) but the effect curiously depended on cell density. In other words, the mutant appeared to grow better than wildtype at low cell density but worse than wildtype at high cell density. Between 8 hours and 24 hours, the mutant growth accelerates and becomes faster than wildtype. We scratched our heads for weeks, stumped by the data.
It was a disappointment and frustration for the family to see timelines slip and to encounter confusing results that ground the work stream to a halt. It was also a gut punch to us to not be able to deliver on our promises. Alas such is science at the edge of precision medicine. The only way through an impasse is stubborn creativity. Because we run drug repurposing as a service for multiple families/foundations, we can implement fixes across projects in real-time.
When we ran into a wall trying to make patient avatars, we pivoted to the DAmP strains. When the DAmP strains failed us, we pivoted again. This time to a collection of temperature-sensitive (ts) mutants that was published by Prof Charlie Boone’s lab at the University of Toronto, a powerhouse of yeast genetics for over 20 years.
At the start of the summer, we received the ade13 ts mutant, which harbors the following roster of mutations. None of these mutations corresponds to a pathogenic variant in humans, but we reasoned that what mattered more was being able to induce ADSL/ADE13 deficiency in an otherwise normally metabolizing yeast cell.

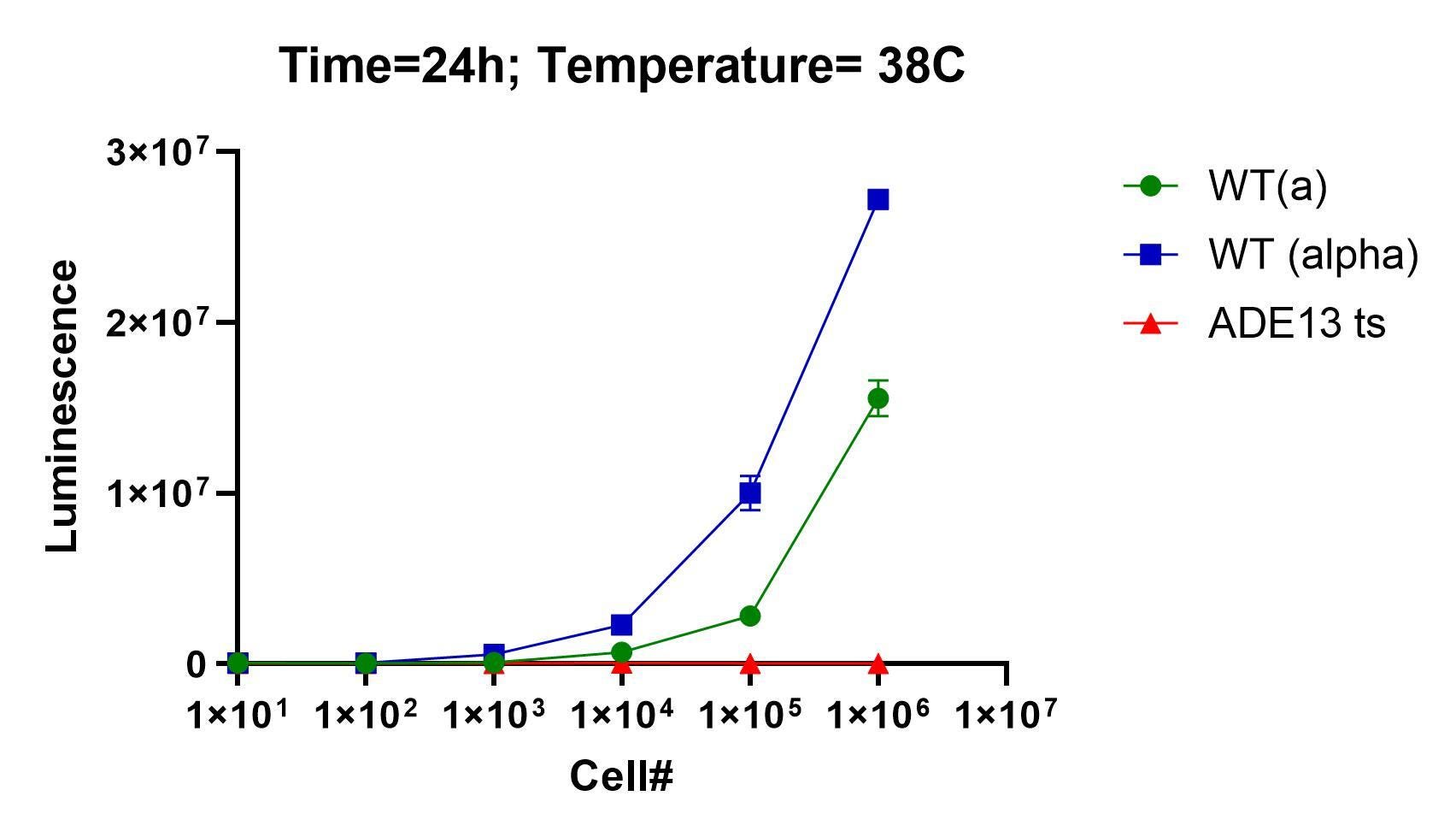
Right out of the gate and to everyone’s relief, the ade13 ts mutant behaved as expected. Here’s the seeding density experiment performed at BADASS labs in late June. It worked on the first attempt.
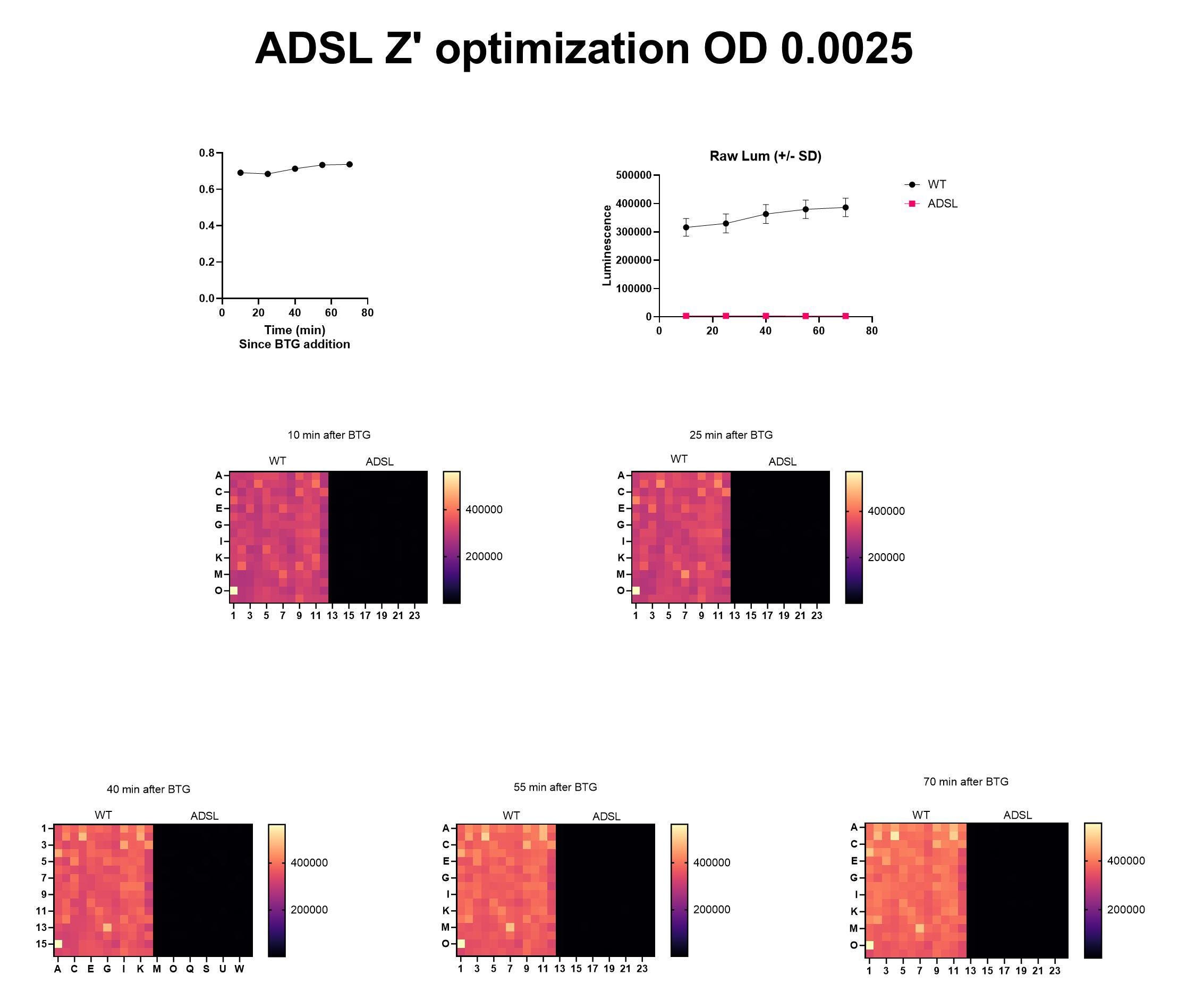
A few weeks later, Natalie and the yeast team at BADASS Labs got the Z’ optimization to work on the first attempt, too.
After months of failure, we were finally hitting out stride over the summer. The Pharmakon screen was finally in sight.
Here’s the ADSL Pharmakon dataset. Presumptive rescuers are shaded in green. Presumptive sensitizers are shaded in red. The negative (-) control is the ade13 ts mutant treated with a placebo (the solvent DMSO). The positive (+) control is a wildtype yeast expressing ADE13. Upon manual inspection of each library plate to eliminate false positives and artifacts, there appear to be seven rescuers and 27 sensitizers that meet the threshold of a hit.

Before we examine the most clinically actionable hit in closer detail, how does ADSL stack up to the other Pharmakon screens we’ve run? ADSL rescuers are difficult to discern because the ade13 ts mutant was quite sick and therefore difficult to rescue. We needed to examine each plate to identify winners and losers.

Now let’s zoom in on one of the Pharmakon library plates that contained one of the top ADSL yeast rescuers: disulfiram.

We have never seen disulfiram come up as a rescuer in any other drug repurposing screen we’ve run to date. If anything, when it’s come up as hit at all, it’s always been a sensitizer! It’s not just the dose that makes the poison. Sometimes, a poison in one disease context is the antidote in another Indeed, with the exception of cetrimonium, each of the sensitizers listed above is a rescuer in a different screen. For example, sodium tetradecyl sulfate was a rescuer in the yeast MEPAN (MECR deficiency) project.
So what the heck is disulfiram, and why do we think it’s the most clinically actionable hit?

Disulfiram was first synthesized by chemists in 1881 for industrial applications. In the 1930s, it was serendipitously discovered to create an acute and unpleasant hangover-like reaction in people who consumed alcohol. In the decades following WWII, the pharmaceutical industry pursued disulfiram as an alcohol cessation therapy, and it was approved by the FDA in 1951, and marketed as Antabuse. Studies of the pharmacology of disulfiram led to the discovery that aldehyde dehydrogenase is the principal drug target. Disulfiram is still available for use in many countries even though it’s been superseded by next-generation treatments for alcoholism.
For obvious reasons, children normally don’t take disulfiram. So it’s not clear what a safe dose would be for a child with ADSL deficiency. There are also open questions about chronic dosing and dosing frequency to which we don’t have answers. No one does because we are at the wild frontier of precision medicine.
Most importantly to point out: we don’t know the mechanism of action by which disulfiram is rescuing ade13 ts mutant yeast cells. Why is aldehyde dehydrogenase inhibition beneficial in the absence of ADSL? Intriguingly, a Google search of the terms “ADSL + disulfiram” reveals a Chinese patent published in 2019 describing a novel use for disulfiram to treat ADSL deficiency. What are the chances?
As next steps in the lab, we hope to perform hit validation experiments in a worm model of ADSL deficiency. Prof Hanna-Rose’s lab recently published on an ADSL deficiency worm model, which manifests disease phenotypes (like embryonic lethality) that can be rescued by drug treatment. We’re also considering redoing the Pharmakon screen at at higher seeding density and slightly lower temperature (37˚C versus 38˚C) in order to make the ade13 ts mutant more rescuable. That should reveal more rescuers and give us a better sense of their relative potency.
Onward for Minja and other kiddos around the world with ADSL deficiency.