CACNA1A Cure Roadmap
The 5th Cure Roadmap was the first one commissioned by a highly motivated parent-led foundation called CACNA1A Foundation.


CACNA1A-related disorders Cure Roadmap
Prepared for CACNA1A Foundation by Perlara PBC
Q1 2022
VISION
Toward a future where all people with CACNA1A-related disorders live healthy, fulfilled, and complete lives. Essential capacity-building work already being supported by the CACNA1A Foundation will deliver a more complete characterization of genotype/phenotype relationships across the patient population, will lead to new in vitro and in vivo disease models for use in elucidating CACNA1A pathophysiology and testing treatment options, and will evaluate a single drug repurposing candidate in a mouse model. Building on top of this impressive base of ongoing research activities, the CACNA1A-related disorders Cure Roadmap aims to guide the CACNA1A Foundation toward efficient and variant-inclusive paths at every step from basic research to clinical development of novel treatments. This ambitious strategy will be supported by performing Cav2.1 in vitro assay development, high-throughput CACNA1A variant analysis and unbiased drug repurposing screens across a network of academic, industry and for-hire laboratories. The resulting therapeutics-enabling datasets will be accessible via an online portal that also links to an expanding CACNA1A biobank and a definitive CACNA1A natural history study that will inform selection of quantitative reversible biomarkers and functional outcome measures for clinical trials.
This roadmap envisions the following milestones and timelines. Within the next 1-2 years, the priority will be to establish new avenues, and amplify existing ones, for small molecule drug discovery and development. We call attention to the need for development of high-throughput, cell-based, functional readouts (assays) of Cav2.1 voltage-gated channel opening and channel conductance, as well as intracellular trafficking of the Cav2.1 channel. Targeted treatment options will be identified from the generation of an open source CACNA1A variant cell line library, constructed in a stepwise hypothesis-driven fashion, that will eventually assess the effects of over a thousand CACNA1A variants in standardized, multiplexed, scalable Cav2.1 functional assays. Data generated from these preclinical proof-of-concept experiments will form the basis for seeking potentially transformational drug development partnerships with industry. Starting with drug repurposing compound libraries and a curated panel of representative CACNA1A-variant-expressing cell lines, a focus on small molecules is currently the approach most likely to rapidly deliver novel therapeutic options for the largest number of CACNA1A patients.
The CACNA1A Foundation is in the best position to implement learnings from the Cystic Fibrosis Foundation experience which involved a two-decade venture philanthropy partnership with Vertex Pharmaceuticals that resulted in development of blockbuster small molecule medicines like the triple-combination CFTR conductance corrector therapy Trikafta that today treats 90% of CF patients. The proceeds of small molecule medicines commercialized since 2012 have not only saved thousands of lives and created billions of dollars in company value, but are also supporting development of genetic therapy options to fix the long tail of 10% of CF patients with private or pharmacologically intractable mutations. This roadmap recommends going full steam ahead on small molecules over the next two years and building the research capacity to achieve precision pharmacology for CACNA1A.
Within the next 2-5 years, as minimally invasive and ultimately non-invasive brain delivery technologies emerge, the CACNA1A Foundation’s focus will expand to include avenues for developing prime editing (or successor technologies), which is a genetic therapy that has the potential to be curative, requiring only a single dose, and broadly applicable to the majority of CACNA1A patients, especially for patients with variants that turn out to be pharmacologically intractable. Antisense oligonucleotides are a genetic therapy modality that will be in play much sooner for classes of amenable variants and they offer the potential to treat multiple segments of the CACNA1A-related disorders patient population. The CACNA1A Foundation will begin laying the framework for these precision and even individualized therapy approaches immediately, by establishing relationships with key scientific leaders from academia, nonprofit and biotech in the area of utilizing genetic therapies for central nervous system (CNS) disorders.
By year 3 (2024), we envision that the CACNA1A Foundation will be actively supporting a $1M annual research budget with milestone-driven, translation-focused research grants and expanding into high-throughput preclinical testing capabilities in the years after. By year 5, data from a definitive natural history study combined with data from two or more Phase 1/2 studies will form the backbone of an emerging portfolio of mid-to-late-stage clinical assets. Over the next 5-10 years, multiple industry partners will have clinical-stage programs for CACNA1A-related disorders and these partners will either be in co-development collaborations with the CACNA1A Foundation or the CACNA1A Foundation will have out-licensed wholly or partially owned assets to industry partners in exchange for equity and/or royalties. Three or more medicines for CACNA1A-related disorders are forecast to be approved by the end of this decade.
AUTHORS
Ali Althaus, PhD (Perlara Cure Guide)
Sej Chung, PhD (Perlara Cure Guide)
Sam Finlayson, MD, PhD (Perlara Cure Guide)
Ethan O. Perlstein, PhD (CEO of Perlara PBC & Maggie’s Pearl LLC)
EXECUTIVE SUMMARY
The CACNA1A Cure Roadmap lays out a strategy for community-supported and variant-inclusive drug development with the ultimate goal of identifying novel treatments for all patients with CACNA1A-related disorders within a decade. The near-term recommendations of this roadmap are for the CACNA1A Foundation to support Cav2.1 functional assay development, CACNA1A variant characterization, and electrophysiology-based drug repurposing with an eye toward scalable small molecule drug discovery and development that will safely and effectively treat the majority of the mutationally diverse CACNA1A patient community. The long-term vision incorporates one-time genetic therapy approaches which are promising conceptually but require further breakthroughs in delivery of therapeutics to critical brain circuits that are still several years off in the future, as well as multi-year safety studies.
Specific goals include screening the Broad Repurposing hub compound library to identify Cav2.1 activators that normalize channel function in a haploinsufficient CACNA1A cell line; Cav2.1 inhibitors that selectively normalize channel function in a cell line expressing a well-characterized CACNA1A gain-of-function mutation; Cav2.1 inhibitors that selectively normalize channel function in a cell line expressing a well-characterized CACNA1A dominant-negative mutation; generating an initial library of Cav2.1 channel variants for multiplexed cell-based functional assay development that will group mechanistically similar CACNA1A variants into discrete classes.
It will quickly pay dividends to make the upfront investment to classify CACNA1A variants in bulk, and make these classifications open and accessible via an online portal, rather than individual labs performing one-off and data-siloed functional assessments of novel variants of unknown significance as new families are diagnosed around the world. Patients will ultimately be grouped according to the functional impact of their individual variants and matched with targeted treatment options. Ideally a few treatment options will benefit most patients with CACNA1A-related disorders. Long-term directions include exploring genetic therapy strategies, initially antisense oligonucleotides and eventually a universal gene editing platform technology like prime editing, which has the potential to house a single, all-inclusive, flexible therapeutic approach that could fix the root cause of the pathophysiology of CACNA1A-related disorders in a totally variant-agnostic manner. However, the CACNA1A Foundation should note that realizing the promise of safe, one-time, corrective gene editing in the deepest parts of the brain will require technological breakthroughs in therapeutics delivery that are impossible to predict today. Antisense oligonucleotides (ASOs) are a genetic therapy modality that can be applied to certain classes of CACNA1A variants as well as in individual cases due to amenable private mutations.
The most variant-inclusive and brain-penetrant therapeutic modality with proven track records of safety and deliverability are without question small molecules. The CACNA1A community is well-positioned for rapid small molecule therapeutic advancement. Although some critical gaps remain like a solved Cav2.1 protein crystal structure, decades of research have documented the primary function of the CACNA1A protein, the voltage-gated calcium channel Cav2.1, and its role in human disease. Recent advancement in the efficiency and availability of genetic screening have identified novel patients with CACNA1A mutations and expanded the spectrum of known CACNA1A-related disorders.
A multi-species array of cell and animal models of CACNA1A-related disorders have been published and numerous clinical and scientific experts are working to generate and characterize additional disease models to better understand and represent this heterogeneous patient population. An ongoing natural history study and patient data collection program may provide foundational knowledge to inform clinical trial endpoints and may potentially be used as comparison data for therapeutic efficacy.
The CACNA1A Foundation has assembled a formidable coalition of patients, families, researchers, and clinicians united around an inclusive mission of identifying treatments and cures for all CACNA1A-related disorders patients. In addition to endorsing ongoing research and capacity-building activities, this roadmap calls for three Year One (2022-2023) community-funded research projects that will benefit all CACNA1A families, increase the visibility of CACNA1A-related disorders, and catalyze conversations with biotech and investors that will culminate in sponsorships and/or partnerships:
Cav2.1 in vitro functional assay development and CACNA1A variant characterization by the deep mutation scanning (DMS) approach in collaboration with an academic lab that offers DMS as a sponsored research project and is looking to partner early with rare genetic disease foundations that would benefit from precision pharmacology. Estimated cost for 12 months of work with an academic partner lab is at least $300,000 to be paid as multiple go/no-go milestone payments tied to contractually defined deliverables.
Unbiased drug screening of a panel of representative CACNA1A-variant-expressing cell lines followed by validation studies in patient iPSC-differentiated neurons to identify novel drug repurposing/repositioning candidates. Estimated cost for 12 months of work with an academic partner lab or contract research lab is $250,000 to be paid as multiple go/no-go milestone payments tied to contractually defined deliverables./
Creation of a user-friendly CACNA1A-related disorders online portal and federated data sharing platform to encourage precompetitive discovery, spark new research collaborations, and facilitate connections with potential industry partners or funders. Estimated cost for 12 months with an academic partner lab is zero as their services will be delivered in-kind or will be supported by external grant funding.

INTRODUCTION
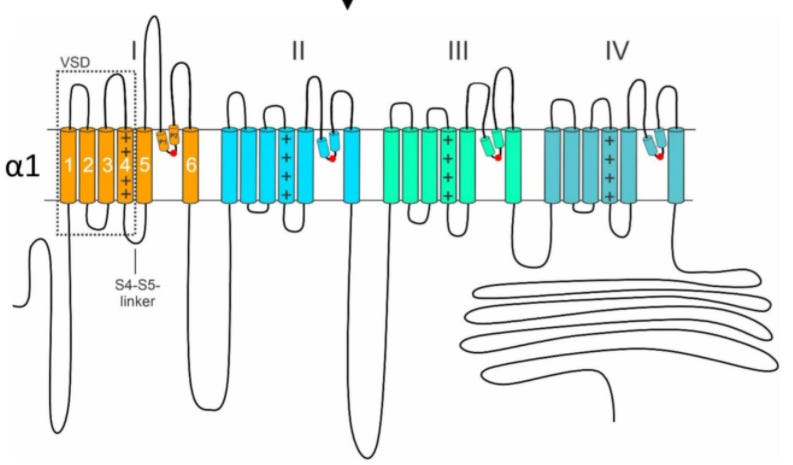
The CACNA1A gene encodes for the α1A subunit of the P/Q type voltage-gated calcium channel (VGCC), also known as Cav2.1. The α subunit of VGCCs forms the ion conducting pore and assembles with one β, one γ, and one α2δ subunit to form the fully functioning channel. The P/Q type channel is found primarily on presynaptic terminals, dendrites, and neuroendocrine cells in the brain and it is critical for regulating synaptic transmission, among other functions. In 2013, the gene was found to have bicistronic expression and encode a separate functional protein that shares the sequence of the cytoplasmic C-terminal tail and is called α1ACT. This second protein acts as a transcription factor that is critical for cerebellar development and cell viability, and its expression in wildtype form is sufficient to partially rescue the phenotype of a CACNA1A knockout mouse.
The causal role of CACNA1A in human disease was first identified in both Familial Hemiplegic Migraine (FHM) and Episodic Ataxia Type-2 (EA-2). FHM is a form of migraines with aura and motor deficits; patients experience paroxysmal hemiparesis, aphasia and loss of consciousness even leading to coma, and many will eventually develop cerebellar atrophy. EA-2 is the most common form of episodic ataxia; patients experience paroxysmal ataxia and migraine-like symptoms, nystagmus, and cerebellar atrophy. Patients with EA-2 are most frequently diagnosed in the second decade of life, and patients with FHM are typically diagnosed in the first or second decade of life after the development of the above symptoms. However, most patients have experienced more neurodevelopmental changes or deficits much earlier, such as delayed motor or language milestones, ADHD, or autism spectrum disorder. Six individual variants were initially reported: four from FHM patients that were missense gain-of-function (GOF) mutations and two from EA-2 patients that were truncating and likely the subset of loss-of-function (LOF) mutations that are dominant negative and impinge or poison wildtype channels.
Dozens of CACNA1A variants have since been published and connected to heterogeneous neurological disorders – and it is reasonable to assume that hundreds if not over a thousand CACNA1A variants will at some point be observed in patients. While additional variants have been identified in patients with FHM and EA-2, they remain primarily associated with missense GOF mutations and truncating, presumably dominant-negative LOF mutations, respectively. Spinocerebellar Ataxia 6 (SCA6) is a late-onset progressive cerebellar ataxia which is most frequently associated with an expanded CAG(n) repeat. SCA6 has also been linked to dysfunction of the α1ACT protein. The role of this protein in other CACNA1A-related disorders is currently unknown, and is being actively investigated in Dr. Christopher Gomez’s lab with a grant funded by the CACNA1A Foundation. For that reason, α1ACT is not clinically actionable in Year One (2022) but a line of sight is visible for an α1ACT gene therapy vector design and proof-of-concept safety and efficacy experiments in a mouse model in Year Two (2023).
However, strong evidence demonstrates a key role for α1ACT in developmental programming of the cerebellum, which may therefore have implications for other disorders such as FHM and EA-2. Novel GOF and LOF mutations have also been established in patients with Developmental and Epileptic Encephalopathy 42 (DEE42) which is characterized by the onset of various refractory seizures as early as the first hours of life, associated global developmental delay and impaired intellectual development, as well as axial hypotonia, peripheral hypertonia, tremor, ataxia, and abnormal eye movements. DEE42 accounts for approximately 1% of patients in the large Epi4K epilepsy consortium. Additionally, patients with CACNA1A variants may present with numerous symptoms associated with each of these individual disorders, rather than fitting within one particular subgroup.
CACNA1A-related disorders are typically autosomal dominant and, with the exception of SCA6, present early in life. While there is marked clinical heterogeneity both between and within CACNA1A-related disorders, general temporal patterns have been identified, with epileptic encephalopathy usually presenting within hours or days of birth, and episodic ataxia typically presenting later in life. Of note, Indelicato and Boesch comment that even patients that present with severe symptoms later in life tend, upon further inspection, to have exhibited a mild intellectual impairment from a young age. A major natural history study organized by Dr. Wendy Chung and the CACNA1A Foundation is in progress and will provide more detailed information about the disease course across patients. This work is already yielding important insights about the range of neurological symptoms that can be associated with CACNA1A disease-causing variants and the most commonly used and most effective medications.
This roadmap endorses the six ongoing research activities and proposes two new research activities, as summarized in the bullet points below:
Partnership with Dr. Wendy Chung at Columbia University on a Natural History study which will provide in-depth, longitudinal data about the CACNA1A patient experience and disease presentation and progression that can help identify how the patient experience varies by genotype. Ultimately, data from this study will lead to better-informed treatment options for individual patients and may also be used to inform clinical trial designs.
Partnership with RARE-X on a data collection program to provide additional insight on genotype/phenotype relationships within CACNA1A patient experiences, which will be critical to designing patient-reported outcomes (PROs).
A funded research grant supporting the work of Dr. Annapurna Poduri and Dr. Ingo Helbig to characterize epilepsy-related phenotypes of CACNA1A patients to better understand genotype/phenotype relationships among CACNA1A patients that experience seizures, and to determine the functional effects of CACNA1A variants in these patients.
A funded research grant supporting the work of Dr. Henry Colecraft and Dr. Christopher Gomez to generate 10 novel patient-derived induced pluripotent stem cell (iPSC) lines and investigate the role of both Cav2.1 and α1ACT on phenotype in neuronal cells.
A three-year contract with CombinedBrain and ibx to develop an unlimited number of iPSC lines from patients at a discounted rate.
A CACNA1A data portal being created by Dr. Dennis Lal’s group to be eventually populated with the DMS and drug repurposing datasets described below and that is similar to the GRIN Portal at the Broad Institute. This CACNA1A data portal can serve as a centralized hub for relevant data being generated across these and other experiments being done by CACNA1A/Cav2.1 researchers throughout the scientific and medical community.
Cav2.1 in vitro functional assay development and generation of an initial CACNA1A variant cell line library using the deep mutational scanning (DMS) approach in collaboration with an academic partner lab to study the effects of many variants on Cav2.1 protein function and to use this dataset to inform multiplexed high-throughput drug screening and other applications.
Unbiased drug repurposing screens using a panel of up to 5 CACNA1A-variant-expressing cell lines and electrophysiological readouts to be performed by an academic core facility or contract research organization like Charles River Laboratories, followed by hit validation studies in patient iPSC-differentiated neurons to be performed by an academic collaborator.
In parallel with the efforts in progress to better define the CACNA1A patient population and experience, this roadmap proposes a multi-stage translational research program designed to rapidly identify promising drugs for repurposing and to explore genetic therapy options with the following caveats in mind. Given the dosage sensitivity risk of CACNA1A overexpression and the infeasibility of a CACNA1A “mini-gene” to circumvent the payload limits of current vector-based gene delivery technologies, genetic therapy options should focus on reversible genetic therapies like antisense oligonucleotides, or ASOs. Splice-modulating or exon-skipping ASOs could work for certain types of LOF mutation; RNaseH-sensitive knockdown ASOs could work for classical GOF mutations; allele-selective ASOs could work for certain GOF mutations; and gene-activating ASOs could work for all classical (non-dominant-negative) LOF mutations. Emerging small molecule platform technologies like brain-penetrant targeted protein degraders (TPD) and targeted RNA degraders (TRD) hold the promise of replacing intrathecally delivered ASOs with equally efficacious oral pills.
Based on our assessment of the CACNA1A knowledge base as it exists today, we recommend the following high priority translational research objectives which we believe are on the critical path to an approved CACNA1A small molecule therapeutic: (1) developing assays that measure Cav2.1 electrophysiological activity, intracellular trafficking and activity-dependent localization; (2) classifying CACNA1A variants into pharmacologically similar groups, starting with many possible missense mutations as well as known variants of unknown significance including frameshift, indel, nonsense and non-coding mutations; (3) exhausting cell-based drug repurposing/repositioning using a small panel of hand-picked, exemplar CACNA1A variants and standardized electrophysiological channel activity readouts; (4) expanding the CACNA1A iPSC biobanking and deep phenotyping effort so as to obviate the need to demonstrate functional efficacy (as opposed to target engagement) in a mouse or rat model.
Further, there are open questions that may be answered with $25,000 to $50,000 RFP and grant funding opportunities. One of the biggest unanswered basic science questions is in regards to the crystal structure of Cav2.1. Solving this structure will unlock both in silico and mechanism-based drug discovery efforts in academia and industry. Another unanswered scientific question is the translational value of invertebrate (worm and fly) CACNA1A disease models. The low upfront cost and high upside potential of worm and fly CACNA1A patient avatars make these experiments worthwhile. A clinical trial preparedness question that needs to be addressed is: what is the strategy for biomarker and clinical outcome assessment and how will public-private partnerships between CACNA1A Foundation (and other allied nonprofits) and industry or investors play a role? Grants can also allow for investigation into the consequences of CACNA1A mutations on cerebellar circuits in mouse models, which could reveal targeted treatment options inaccessible to in vitro approaches.
THERAPEUTICS READINESS
Similar to the Cystic Fibrosis Foundation experience, a key challenge for small molecule drug development for CACNA1A-related disorders is a complex genetic architecture involving a large number of variants, in particular de novo variants. Adding further complexity is the fact that CACNA1A variants exhibit distinct genetic mechanisms spanning classical loss-of-function variants, classical gain-of-function variants, as well as non-canonical variants. Therefore, a unique challenge faced by the CACNA1A Foundation is determining the scope and distribution of variant functional effects across the entire CACNA1A-related disorders patient community.
The goals of the experiments proposed in this section of the roadmap are twofold: (1) empirically establish genotype/phenotype relationships across the spectrum of possible CACNA1A variants, including documented variants of unknown significance; (2) begin to define potential therapeutic subpopulations based on cellular response to repurposable compounds. The deep mutational scanning and drug repurposing screen experiments can be done sequentially, such that drug repurposing builds on the tools (cell lines) and knowledge (variant effects) gained from deep mutational scanning. Alternatively, they can be done in parallel, with drug repurposing screens being executed using a small sample of representative variant cell lines generated expressly for that purpose.
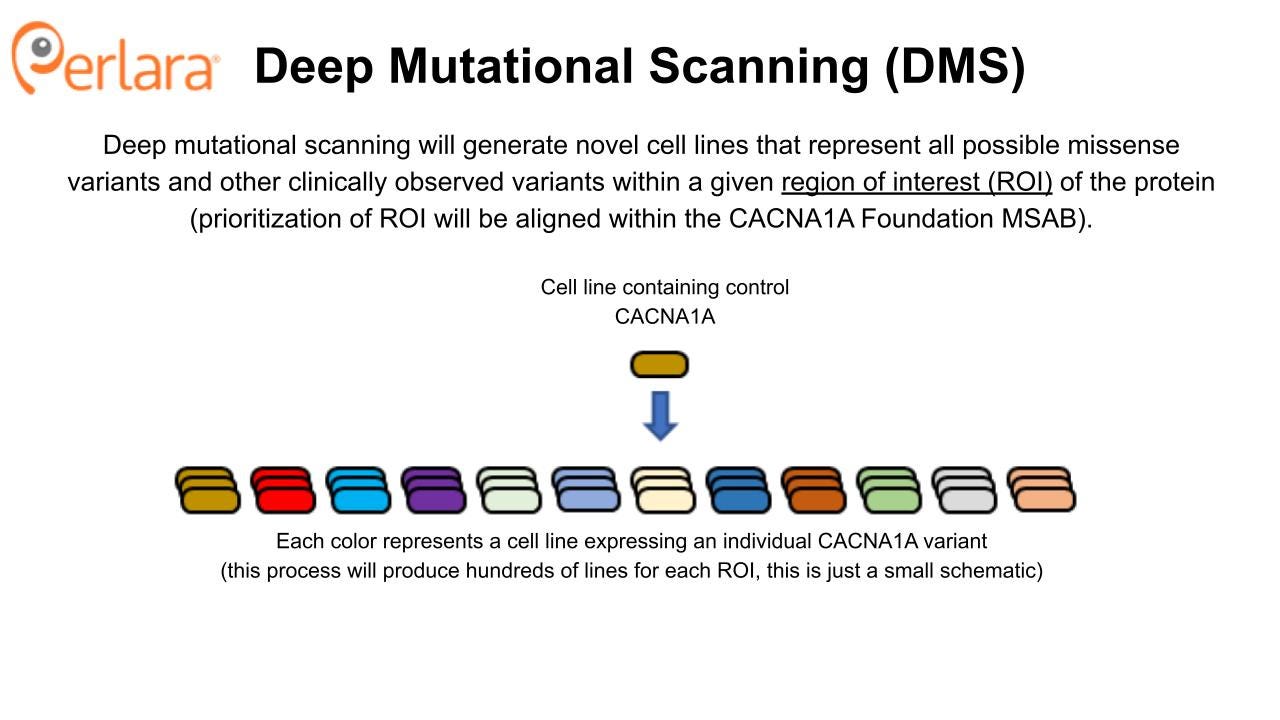
CACNA1A Deep Mutational Scanning
The approach to screen for repurposable compounds that impact wildtype and representative mutant Cav2.1 channel function maximizes efficiency; potential therapeutic compounds can be identified and tested against validated disease models within the span of one year. However, we acknowledge that this variant-by-variant approach to compound screening, whether drug repurposing or drug discovery, will not be practical or successful for targeting all CACNA1A mutations. Additionally, for some variants, having a compound that selectively targets the mutated protein as opposed to the wildtype protein may be necessary. To address the need for small molecule therapeutics that target groups of mechanistically related CACNA1A variants, and in lieu of assessing variants one at a time in a distributed fashion, a comprehensive variant cell line library will be generated using the deep mutational scanning (DMS) approach as described in Jones et al., 2020.
Although some variants may be classified as LOF or GOF based on 3-D homology structure modeling or simple bioinformatics predictions, the DMS approach offers a proactive, highly efficient solution to functional classification of CACNA1A variants instead of a reactive, ad hoc approach. CACNA1A variants or deletions resulting in haploinsufficiency do not require explicit modeling as in the case of nonsense mutations and frameshift mutations leading to premature stop codons (10-15% of CACNA1A-related disorders patients) and can be modeled by a generic CACNA1A+/- heterozygous deletion cell line. The steady drip of de novo variants of unknown significance creates a bottleneck on all subsequent steps of therapeutic development.
DMS takes advantage of advances in high-throughput genetic sequencing and protein display to identify and evaluate huge numbers (>10^5) of genetic variants in a relatively small number of experiments. The CACNA1A protein is 2506 amino acids long and there are 19 possible amino acid substitutions per position, yielding 47,614 possible missense variants of the CACNA1A protein. However, we recommend a phased, iterative approach to library construction based on the guidance of the CACNA1A Foundation Medical and Scientific Advisory Board (MSAB). Once generated, the cell line library will become a foundational tool that can be utilized for future characterization and drug screening experiments, in addition to those conducted with DMS.
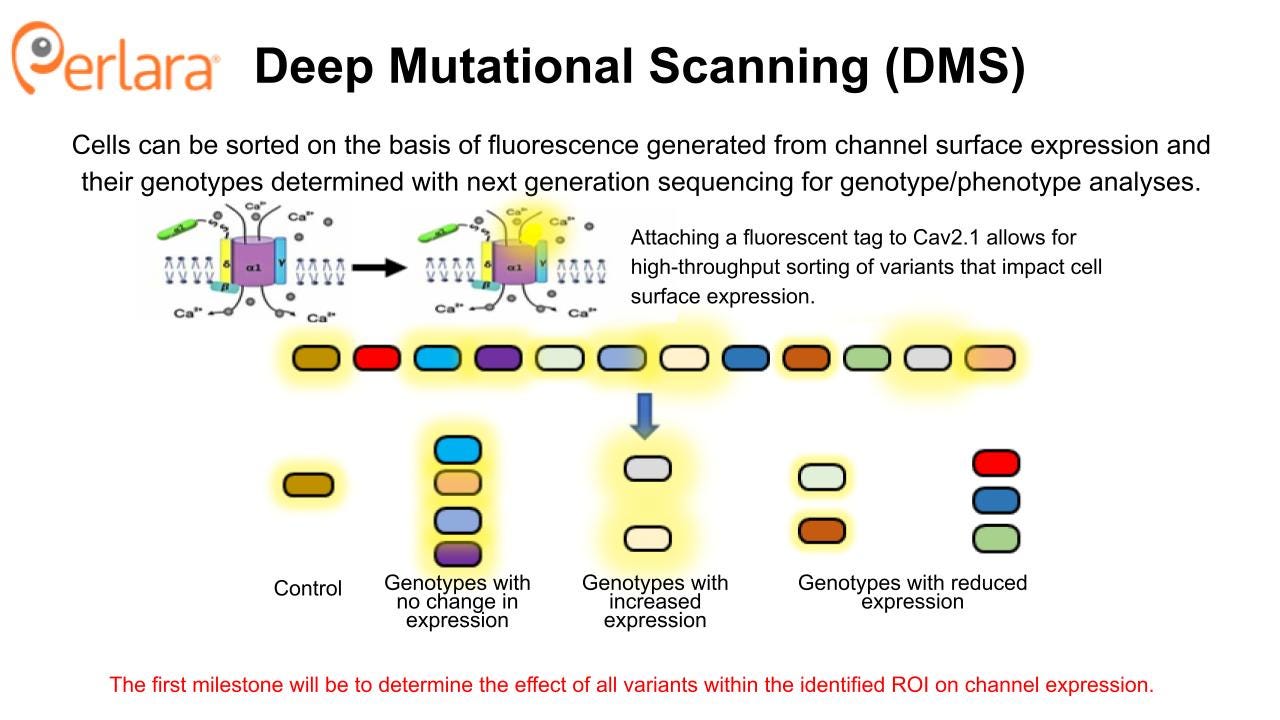
This approach to cell line generation provides an economy of scale and allows for direct comparison of results obtained across different variant lines, thus offering an advantage to piecemeal generation of cell lines or experiments done using transient protein expression in individual laboratories. Furthermore, this rationally constructed CACNA1A variant cell line library will be a complementary resource to the CACNA1A Foundation’s patient-derived iPSC biobank. From a practical standpoint, experiments on iPSC-differentiated neurons are expensive, time-consuming, and require independent laboratory replication. Not to mention costs increase linearly with the number of lines. So a CACNA1A variant cell line library will serve as an economical proving ground for functional assays that will be deployed in patient-derived IPSC-differentiated neurons – or even brain organoids.
This work will be accomplished through a partnership with an academic group that has performed DMS in the context of a different channelopathy and that is willing to perform the experiments at cost.The DMS approach has recently been applied to characterizing variant effects on ion channels. For example, Glazer et al., 2020 describes the application of DMS to the voltage sensor region of the voltage-gated sodium channel gene SCN5A and a biorXiv preprint (Coyote-Maestas et al 2022) was posted during the review of this roadmap by the CACNA1A Foundation MSAB that describes the application of DMS to the Inward Rectifier K+ potassium channel Kir2.1.
The Coyote-Maestas preprint lays out the rationale for why DMS is an attractive approach for CACNA1A and a general template for the experiments the CACNA1A Foundation wants to do to characterize the cell line library. While encouraging, neither the Kir2.1, nor the SCN5A example can be used as a ‘template’ for the DMS experiments needed to fully characterize variant effects for CACNA1A. For example, there are a several features that make Kir2.1 a simpler target than Cav2.1 for the DMS approach:
1) Cav2.1 is larger than Kir2.1 (2506 vs 427 total amino acids). However, this concern should be surmountable, especially with an iterative approach, creating a series of smaller libraries that focus on certain parts of the protein (like the Glazer et al. 2020 listed above).
2) Cav2.1 requires auxiliary subunits for functional assembly at the membrane. An untested question that will be answered in assay development is whether auxiliary subunits complicate the execution of the screen, but the fact that subunit identity can impact trafficking and function may limit the generalizability of the data.
3) Cav2.1 is voltage-gated – this will require more creative assay development for a functional imaging assay, but it should be surmountable using techniques developed for other Cavs (e.g., co-expressing a K+ channel or using gramicidin and media lacking sodium (as in the Belardetti et al., 2009 example) and then modifying K+ concentration to stimulate a voltage shift.
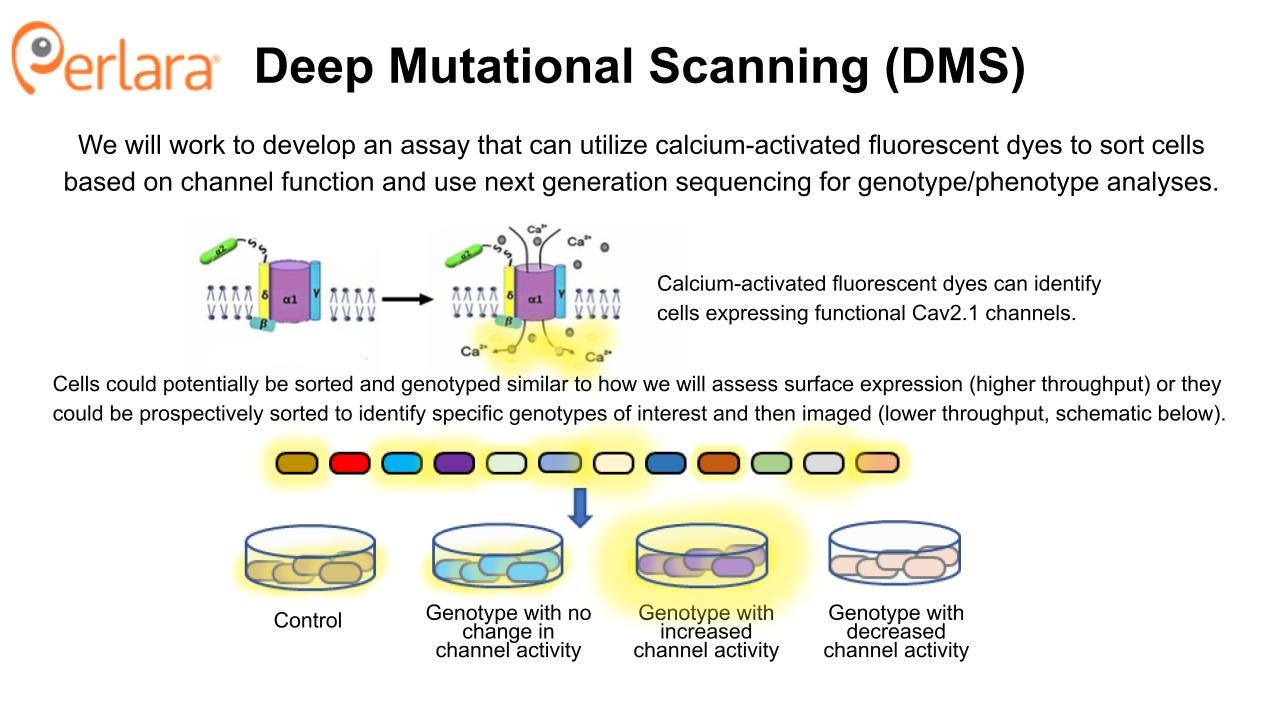
The most straightforward experiments to execute will be those designed to identify protein folding and trafficking deficits and then screen for compounds that correct these deficits. While the distribution of variant functional effects is not fully characterized, a number of LOF mutations have been reported to result in reduced Cav2.1 channel trafficking to the cell surface and retention of the protein in the ER (see, for example: Jeng, et al., Wan, et al., Jiang, et al.). Therefore, these experiments are a complementary approach to the high-throughput electrophysiology-based drug repurposing screen that will be described in the next section, and provide an opportunity to identify novel small molecules that can target a different aspect of the pathophysiology of CACNA1A-related disorders.
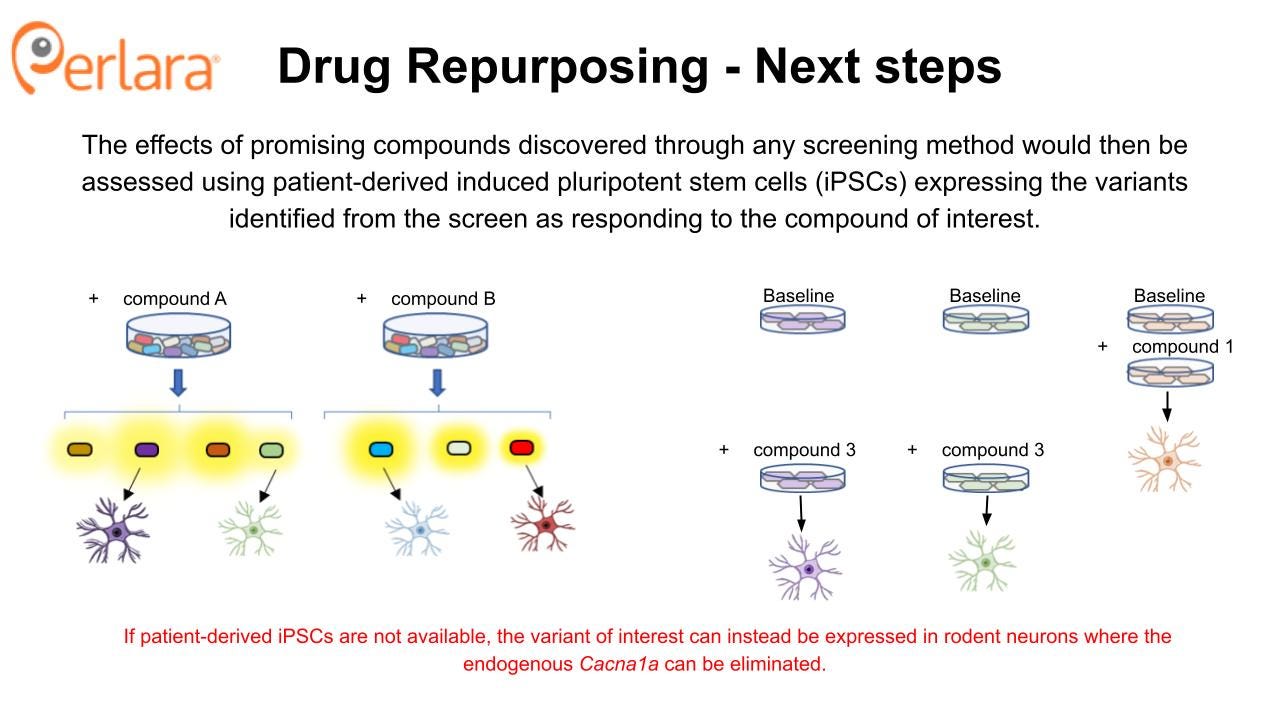
Compounds that are found to increase cell surface expression CACNA1A protein using a corrector screening strategy will then be assessed for ability to impact protein surface expression and function in neuronal cell models expressing the targeted variants. In principle, one or more novel small molecule correctors that rescue multiple CACNA1A LOF variants will be discovered. These validation experiments can be done using patient-derived iPSC lines from patients with matching variants, starting with the first 10 patient lines being established in the labs of Dr Colecraft and Dr Gomez. Alternatively, if such iPSCs are not available, this can be done by transiently expressing the variant in rodent neurons where the endogenous Cacna1a can be eliminated. The F1406C mouse model carries a knock-in LOF mutation in CACNA1A that has been previously shown to result in reduced cell surface expression and ER retention. The mouse model did not exhibit an overt behavioral phenotype, but there was a significant reduction in calcium currents in Purkinje neurons of these mice. Thus, there may be an opportunity to evaluate the in vivo efficacy of candidate compounds using this model.
The DMS approach provides an opportunity to generate a CACNA1A variant cell line library that will become a foundational tool for comprehensively characterizing variant effects on channel expression and function, and that can be used to screen for potential new drugs. It can be rapidly applied to identify novel treatments for patients with LOF variants that impact protein folding and trafficking. However, these mechanisms are believed to represent a minority of patients, so establishing an assay or set of assays that can characterize functional impact of GOF and other LOF variants is critical for full classification of CACNA1A variants using these cell lines.
Electrophysiology assays are the gold standard for characterizing effects on ion channel currents, but are relatively low throughput compared to DMS. Still, a high-throughput electrophysiology approach could be applied strategically, as described in Glazer et al., 2020, using selected cell lines from the previously generated cell line library. Fluorescence-based assays using a calcium-sensitive dye and fluorometric imaging are higher-throughput than electrophysiology and some have already been designed that address the challenge of working with voltage-gated channels in such assays, as in the Belardetti et al., 2009 example described above. This would still require strategic identification of variants of interest, as the throughput does not match that of DMS approach. Coupled with the availability of a variant cell line library, these approaches would allow for characterizing variant effects on a larger scale than is currently available.
For executing DMS cell line generation and protein expression experiments we would approach the following potential academic partners:
Andrew Glazer and Brett Kroncke, who worked on the DMS screen of the SCN5A voltage sensor in the laboratory of Dan Roden. All are currently at Vanderbuilt University. They have the necessary expertise, but their research focus is more on arrythmia-related ion channels. We will reach out to see if they may still have interest in providing expertise on experimental design or possibly executing experiments.
Doug Fowler is a leader in the DMS space, serves on the executive committee for the Atlas of Variant Effects organization, and is a co-author on the SCN5A voltage sensor manuscript. He would be an excellent resource for helping to guide the experimental design/strategy as well as execute experiments or help identify other potential partners.
For functional assay development to characterize variant cell lines generated for DMS, we would approach the following potential partners:
Roden and Glazer labs (above) for electrophysiology experiments
SB Drug Discovery for electrophysiology and plate-based fluorescence imaging experiments
Charles River Labs Charles River Labs for electrophysiology and plate-based fluorescence imaging experiments
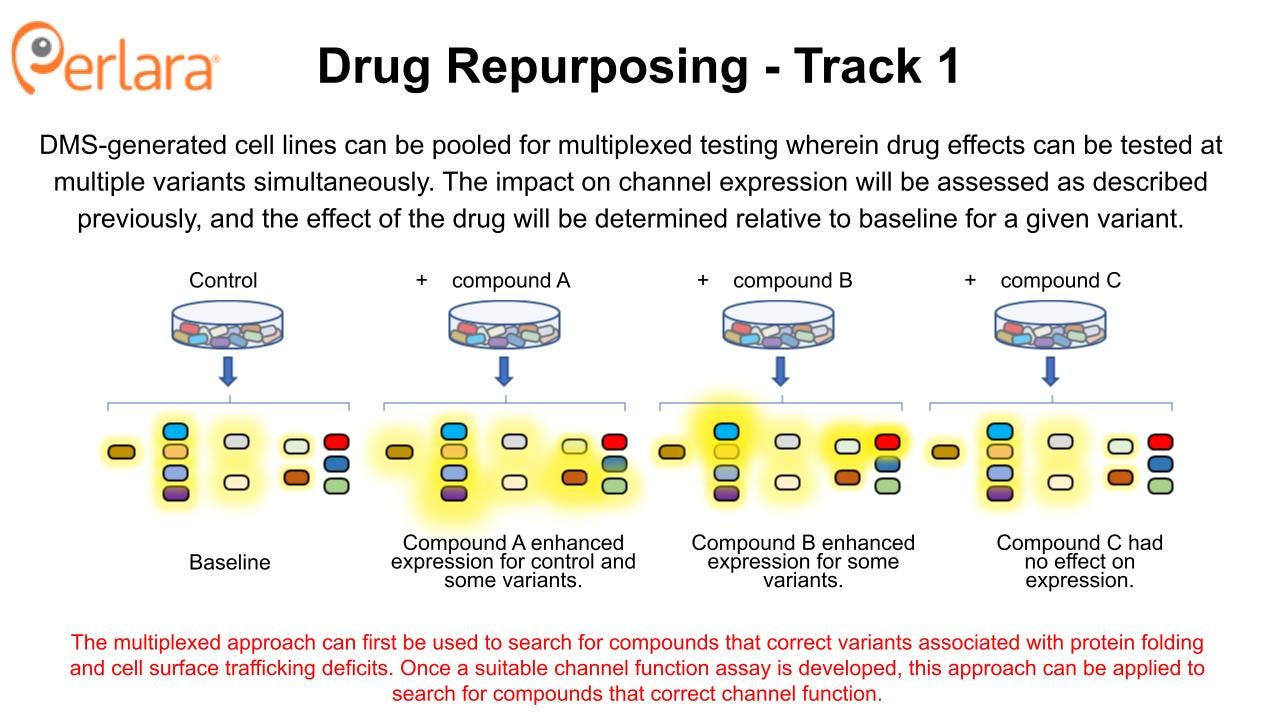
CACNA1A Drug Repurposing 1.0
The goal of drug repurposing/repositioning is to identify a new use for an already approved and marketed drug (repurposing) or a new indication for an experimental drug that has shown early-stage safety, but failed late-stage efficacy studies (repositioning). This approach can be tremendously efficient, significantly reducing the time and cost from discovery of functional effect to patient access if good candidate drugs are identified.
The proposed experiments will be conducted in heterologous cell expression systems that co-express necessary auxiliary or co-assembly proteins, such as HEK293 cells. The screen is designed to identify compounds that directly modulate Cav2.1 function, so establishing activity in heterologous cells represents a “minimum viable product” dataset that can be utilized to seek biotech and industry partnerships and to justify additional cost and time investment required to characterize drug effects in more sophisticated and expensive models like patient-derived iPSCs and in vivo rodent models. Patient-derived iPSCs are a crucial tool for in-depth characterization of variant effects on neuronal function and they will play an important role in establishing efficacy of potential novel drugs and providing preclinical rationale for future clinical trials. However, they are far more resource intensive to use compared to heterologous cells, and are thus not amenable to the type of drug screening being proposed at this stage.

Cell-based electrophysiology screening
The Broad Repurposing Hub (commercially available as the SPECS library) is an ideal launchpad for identifying drug repurposing and drug repositioning candidates. In the first phase of this approach, the SPECS library will be screened on stable cell lines expressing human Cav2.1 channels in HEK293 cells at 100% wildtype or 50% wildtype (i.e., haploinsufficient) protein abundance to look for compounds that interact directly with the protein. The CACNA1A Foundation MSAB will convene to select a representative and well characterized LOF variant, GOF variant and DN variant that could be screened in parallel, pending resource availability. Screening will identify compounds that activate or inhibit the wildtype and several mutant forms of the Cav2.1 channel by measuring effects on calcium currents with patch clamp electrophysiology, which can also be performed at high-throughput using multielectrode array technology. Selectivity of compounds at wildtype versus mutant channels will be assessed. Verapamil and roscovitine may be included as positive controls, or as sensitizers in the event a drug combination screening approach is deemed appropriate.
Promising candidates from the initial screens will then be examined for effects on disease-causing CACNA1A variants to ensure they retain the ability to modulate Cav2.1 in the desired manner and begin to identify subpopulations of patients who may respond to treatment with particular candidate compounds. Most likely, this will be done using transient heterologous expression of disease-causing variants in mammalian cells or Xenopus oocytes which are more efficient to use for these experiments and again provide a minimum viable product dataset that is sufficient to begin to engage with industry partners. However, if the relevant patient-derived iPSC lines are available from the work that the CACNA1A Foundation is doing in partnership with Sampled and CombinedBrain, then we encourage the Foundation to explore assay development opportunities to assess channel function and neuronal phenotypes directly in these cell lines.
Candidate compounds that increase activity at the Cav2.1 channel (but don’t over-activate it) could have therapeutic utility in a variety of patients with LOF variants. Indeed, this is the primary hypothesis for the utility of roscovitine, which increases calcium current by slowing Cav2.1 channel deactivation. The potential for roscovitine to correct in vivo behavioral phenotypes associated with CACNA1A LOF is being investigated in the tottering mouse model by Dr. Qinglong Miao with a grant funded by the CACNA1A Foundation. Novel Cav2.1 potentiators identified from the SPECS library screen, and confirmed to show activity in disease-causing LOF variants, can also be evaluated for in vivo efficacy using the well-characterized tottering mouse model.
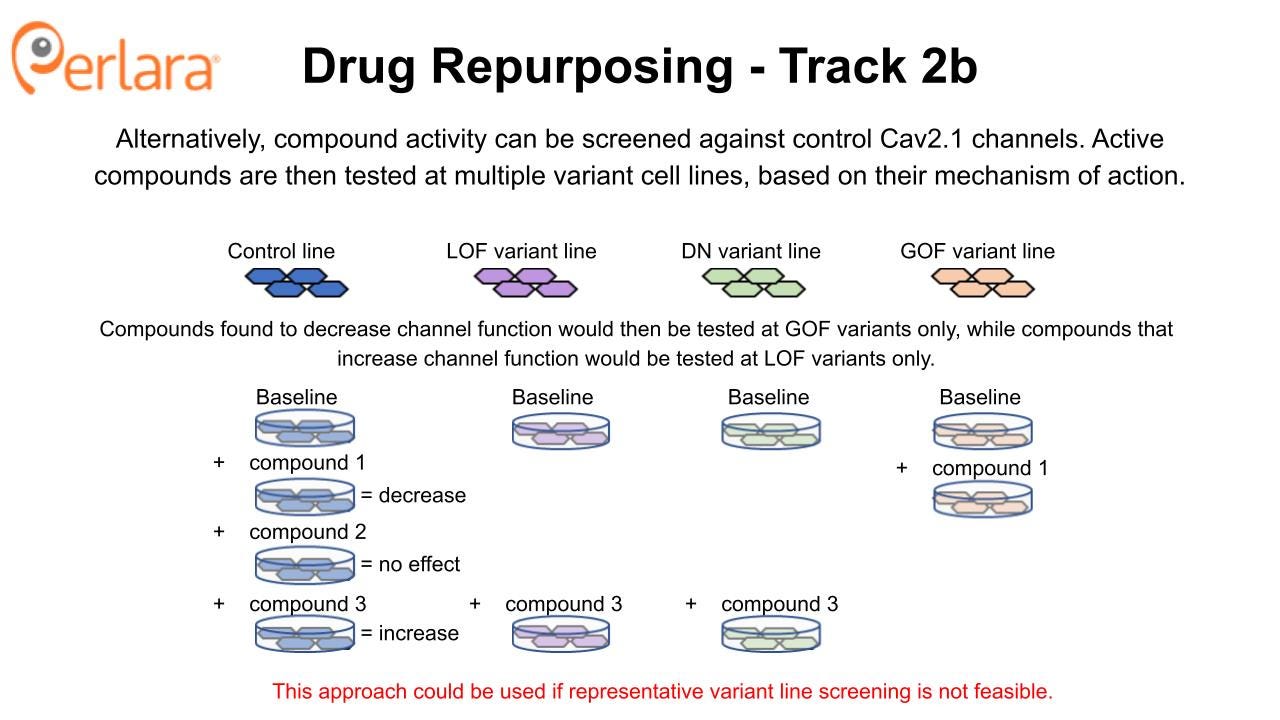
Similarly, candidate compounds that reduce activity at the Cav2.1 channel could have therapeutic utility in a variety of patients with GOF variants. Observed efficacy of flunarizine for treating hemiplegic migraine may be due, in part, to its action as a Cav2.1 channel blocker (it also inhibits other voltage-gated calcium channels). Novel or previously unappreciated Cav2.1 inhibitors identified from the SPECS library screen, and confirmed to normalize activity in disease-causing GOF variants without causing haploinsufficiency, could be evaluated for in vivo efficacy with one of the available GOF knock-in mice. Interestingly, roscovitine also may have therapeutic potential for correcting phenotypes associated with CACNA1A GOF variants because it can antagonize presynaptic calcium channels, as noted by Dr. Miao, and so the compound will also be tested in a GOF model.
These validation studies can be done in partnership with individual laboratories where the models have been characterized, such as with the work being done by Dr. Miao. They can also be done by partnering with biotech contract research organizations that specialize in scalable drug testing in cell and animal models. The decision of where to execute the validation studies will be fit-for-purpose and depend on factors like previous experience with the model, resource availability, cost, and time. Because compounds that modulate Cav2.1 channel function may have therapeutic effects in other patient populations, compounds that show efficacy in CACNA1A variant models may also be tested in other, non-CACNA1A, disease models in preparation for seeking development partnerships.
For electrophysiology HTS screening of the Broad Repurposing library we recommend working with SB Drug Discovery. SB Drug Discovery has a validated Cav2.1 cell line that can be assessed using a Syncropatch (Nanion) automated patch clamp system. They also have capabilities to generate representative variant cell lines for use on this same platform. They are open to exploring cost-sharing opportunities with the Foundation. However, before selecting a CRO we recommend a search and evaluation involving at least three other CROs, especially because there are a plethora of options in the ion channel space.
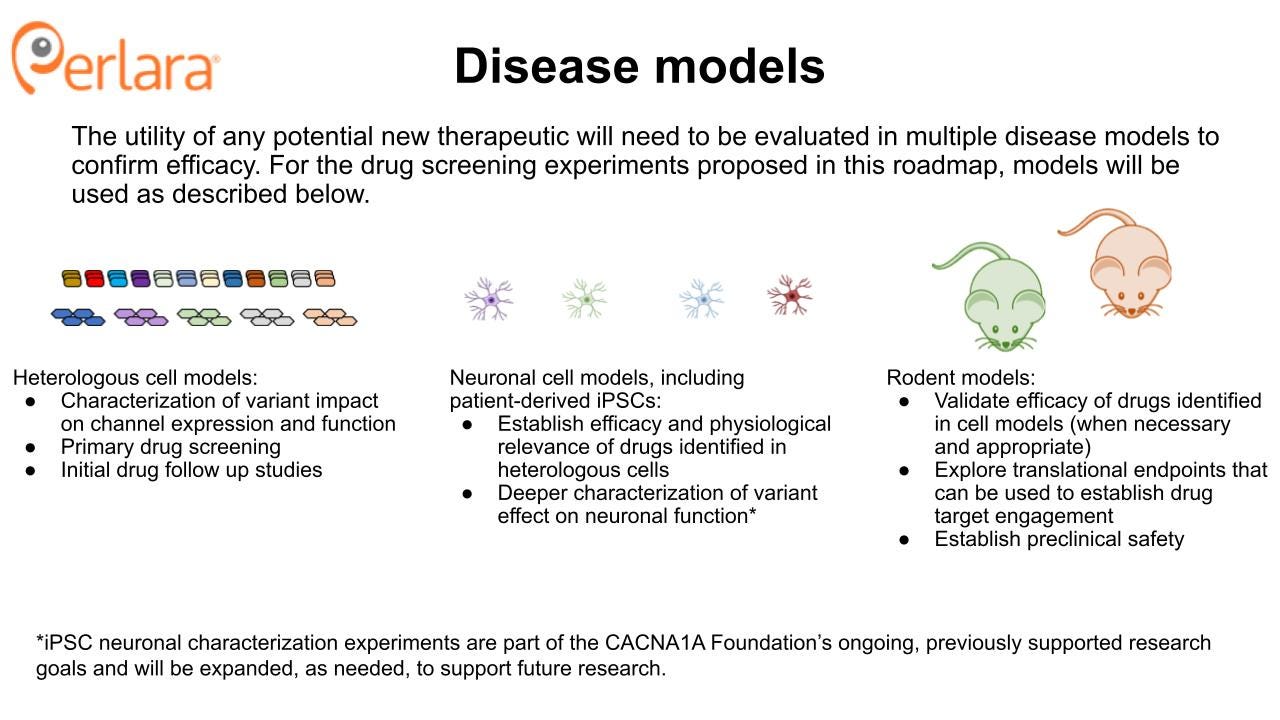
CACNA1A Disease Models
Both of the above approaches focus on screening for compounds that directly impact the biophysical function of the Cav2.1 channel pore. However, the most commonly used pharmacological therapies for CACNA1A patients do not target Cav2.1, and instead modify the system through a different target. This introduces a potential concern that focusing efforts on only targeting the Cav2.1 channel leaves open the possibility that the CACNA1A Foundation could miss therapeutic opportunities that may help restore overall function by working through a different mechanism. For example, pathways that regulate release of calcium from intracellular stores that may buffer or alleviate Cav2.1 hyperexcitability caused by GOF variants. Unbiased phenotypic screening is a technique in drug discovery that can identify compounds that alter a cell or animal model phenotype to promote a desired therapeutic effect, irrespective of the compound’s molecular target.
In order to execute successful unbiased phenotypic screens beyond electrophysiological readouts in immortalized non-neuronal cell lines of the isolated Cav2.1 channel pore, the community needs to establish and validate appropriate cell and animal models and determine clinically and translationally relevant endpoints in said models. CACNA1A plays an important role in synapse function and neuronal excitability, disruption of which may underlie much of the pathophysiology in CACNA1A-related disorders. Modulating the activity of other proteins that interact with CACNA1A in these processes is one approach to ameliorate patient symptoms. For example, this is thought to be why compounds that modulate calcium-activated potassium channels have shown activity in animal models and patients with CACNA1A-related disorders.
Alterations in synaptic function and neuronal excitability have been reported for neurons from numerous animal models and would be expected for many patient-derived iPSCs as well (although not all). High-content imaging techniques have been developed to evaluate related endpoints in large-scale, high-throughput assays that could be amenable to unbiased phenotypic screening. In addition to neuronal cell-based phenotypes, screening at the behavioral level in animals can be a means to identify compounds that have the potential to ameliorate patient phenotypes. This approach would be cost and time prohibitive in mouse models, but zebrafish have been used as models for anticonvulsant phenotypic screening with promising results, and zebrafish models of CACNA1A-related disorders have been developed and exhibit motor and seizure phenotypes.
For CACNA1A-related disorders, a major challenge to phenotypic screening is the availability of scalable models that represent a sufficient proportion of the patient population. For example, only three zebrafish models have been characterized and published, all of which are LOF (though additional models exist for possible use). Available mouse models do represent both LOF and GOF variants but they still only represent a small number of patient variants, and even screening with neuronal cultures from these models would be cost and time prohibitive. Techniques are being developed to improve scalability of iPSC-derived neurons for phenotypic screening, but they will require still more development before they could be applied for sophisticated endpoints related to synaptic function. In addition, few CACNA1A patient-derived iPSC lines have been generated and characterized to date. As more CACNA1A models become available and characterized, and as the genotype/phenotype relationships for CACNA1A patients elucidate more clear therapeutic subpopulations, the unbiased phenotypic screening approach can be periodically refreshed and revisited.
Utilizing cell-based and animal models of CACNA1A will allow the community to identify and validate potential therapies for CACNA1A-related disorders. Fortunately, many such models have already been developed and work is ongoing to generate and characterize others. Once novel drug candidates have been identified from the repurposing/repositioning screens and activity at selected CACNA1A variant channels has been confirmed, the appropriate efficacy validation model(s) will be determined according to: a) relevance to the compound/variant effects, b) availability and accessibility of the model(s) of interest, c) robustness of model phenotypic characterization, d) translational validity (if known), and e) input from potential development partner(s) (if applicable).
Cellular models
Heterologous expression models of CACNA1A variants have been, and remain, crucial models in which to characterize variant effects on properties like protein expression, post-translational regulation, and ion channel physiology. These include both transient and stable expression systems which can also be used to study drug effects. Fortunately, a number of these models have already been studied which represent a spectrum of LOF and GOF disease-causing variants. However, there are many reported variants that remain uncharacterized, which is why this roadmap calls for the generation of a comprehensive library of variants in heterologous cell lines to study their effects and use for drug screening.
Neuronal cell models, particularly those generated from patient-derived induced pluripotent stem cells (iPSCs), are another important cellular model system for studying variant effects. A few such models have been generated and characterized for SCA6 patients. Compared to control iPSC-differentiated neurons, patient-derived iPSC-differentiated neurons did not show differences in expression levels or subcellular distribution of CACNA1A gene products, but they differentially expressed an α1ACT target gene and presented increased vulnerability to cytotoxic stress induced by acute glutamate treatment. These α1ACT and cell viability phenotypes were also described in mouse models of SCA6. To date, no iPSC lines for other CACNA1A-related disorders have been published. Work is ongoing, and supported by the CACNA1A Foundation, to generate and characterize novel patient-derived iPSC lines.
Mouse models
Mouse models of CACNA1A-related disorders predate the discovery of CACNA1A’s role in human disease. Many are spontaneous LOF mutations that have been well-characterized. One example, the tottering mouse model, which exhibits episodic ataxia and absence seizure phenotypes, arose from a spontaneous mutation and was first described in the late 1950’s/early 1960’s. This model, and others like it, provided an opportunity to investigate the pathophysiology of genetic ataxia and epilepsy even before the mutation was localized to CACNA1A. These spontaneous mouse models remain valuable for understanding the role of CACNA1A in biology and pathophysiology and for evaluating the effects of novel therapeutics, especially because their phenotypes have been so robustly characterized. In addition, some of these mouse models are commercially available through The Jackson Laboratory, including the tottering mice.
In addition, the discovery of numerous specific disease-causing CACNA1A variants in multiple patient populations has allowed for the generation of knock-in mouse models that incorporate known disease-causing mutations such as the R192Q model, the S218L model, and the F1406C model. The phenotypes of these models have been characterized with mixed ability to recapitulate features of the human disease. For example, the R192Q FHM-1 model was developed using a gene-targeting approach. This FHM phenotype is closest to that of the common forms of migraine. Functional analysis revealed a pure GOF effect on Ca2+ channel current, neurotransmission, and cortical spreading depression (a phenomenon associated with migraine with aura). These mice have also been found to have sleep disturbances. The S218L mouse model also carries a GOF mutation and has a more severe phenotype than the R192Q; the mice exhibit greater susceptibility to cortical spreading depression, ataxia, and seizures.
The F1406C EA-2 mouse model carries a mutation that was previously known to result in a LOF for the human Cav2.1, and homozygous mice showed approximately 70% reduction in the mouse Cav2.1 current density in Purkinje neurons. Despite this, even the homozygous KI mice did not exhibit an overt motor phenotype, highlighting some of the challenges in translating between animal models and patient data. Nonetheless, mouse models are important research tools and novel mouse models that better recapitulate features and pathophysiology of CACNA1A-related disorders will be useful to evaluate novel treatments, as such mice become available.
Non-mammalian models
Non-mammalian animal models, like zebrafish, Drosophila, and C. elegans can also provide insight into the role of CACNA1A in biological systems and, in some cases, may be amenable for use in high throughput screening. Zebrafish models have been established for numerous genetic epilepsies and a zebrafish model of Dravet Syndrome demonstrated utility in an anticonvulsant phenotypic screen.
In zebrafish, the gene encoding the CaV2.1 α-subunit is duplicated (similar to many zebrafish genes), yielding cacna1aa and cacna1ab. Zebrafish loss-of-function cacna1ab mutants, tb204a and fakir, and a zebrafish partial knockdown mutant have been previously characterized. The tb204a mutation (Y1662N) resulted in reduced motility and synaptic dysfunction at the neuromuscular junction, both of which were reversed by roscovitine. The fakir mutation (L365V) also resulted in reduced motility. The partial knockdown (using microinjections of antisense morpholino oligomers) resulted in epileptiform-like discharges which responded to anticonvulsant drugs.
These zebrafish models are intriguing for their ability to display relevant behavioral phenotypes at a relatively lower cost and higher throughput compared to mice, and thus show some promise as potential future phenotypic screens. However, at this stage, zebrafish are not being considered as a priority model for additional development or investment for this Roadmap. Similarly, while C. elegans and Drosophila have both been used to generate models of CACNA1A mutations, no work is currently being considered in these model systems for this roadmap.
There are useful resources such as ZFIN, which is a zebrafish online database. This database provides ways to access zebrafish with specific mutations in CACNA1A that were inserted into embryos. Included in the list are partial knockdowns that were characterized previously. The cacna1aa orthog in this model is associated with developmental and epileptic encephalopathy, EA2, FHM-1, and SCA6.
CACNA1A is conserved in flies (the Drosophila melanogaster ortholog is called cacophony) and in worms (the Caenorhabditis elegans ortholog is called UNC-2). The higher degree of CACNA1A homology in vertebrates and the established zebrafish disease modeling efforts argue in favor of a laissez-faire approach to invertebrate disease modeling. On the other hand, flies and worms may retain all of the therapeutic predictive value of patient-derived iPSC-differentiated neurons with the added benefit of being whole animal toxicology models. The CACNA1A Foundation should be receptive to funding proof-of-concept drug repurposing screens in flies and worms based on results that are already published.
In 2018, a group published a paper describing fly models of the S218L and R192Q GOF variants. In the past year, another group published a paper describing CACNA1A LOF fly models that display behaviors observed in human patients, including decreased night-time sleep and hyperactivity. In 2016, a group published a paper describing CACNA1A LOF worm models that exhibit rescuable locomotor and behavioral phenotypes reminiscent of phenotypes observed in mouse models. These papers suggest multiple potential opportunities to identify academic collaborators who could be key partners in advancing non-mammalian CACNA1A models for therapeutic screening.

Genetic Therapies
The small molecule approach is fundamentally mutation-agnostic, despite the fact that the comprehensive CACNA1A variant library screen is designed to ensure that even compounds that only show therapeutic potential for a single variant would still be discoverable and actionable. However, a mutation-targeted approach using a one-time genetic therapy has the potential to be curative, provides an alternative to lifelong drug administration (though some families may always prefer a pill), and may be required for some CACNA1A patients with pharmacologically intractable mutations. Antisense oligonucleotides delivered intrathecally to the brain the spinal cord can serve as a bridge between once-a-day oral pills and brain-specific, one-and-done gene editing procedures.
Prime editing is a precise, CRISPR-based gene editing technology that has the potential to correct most disease-causing mutations. It is an emerging, but entirely experimental, therapeutic approach that is rapidly advancing into clinical applications. Despite its promise, significant scientific hurdles remain before prime editing presents a viable option for treating CNS disorders like CACNA1A-related disorders.
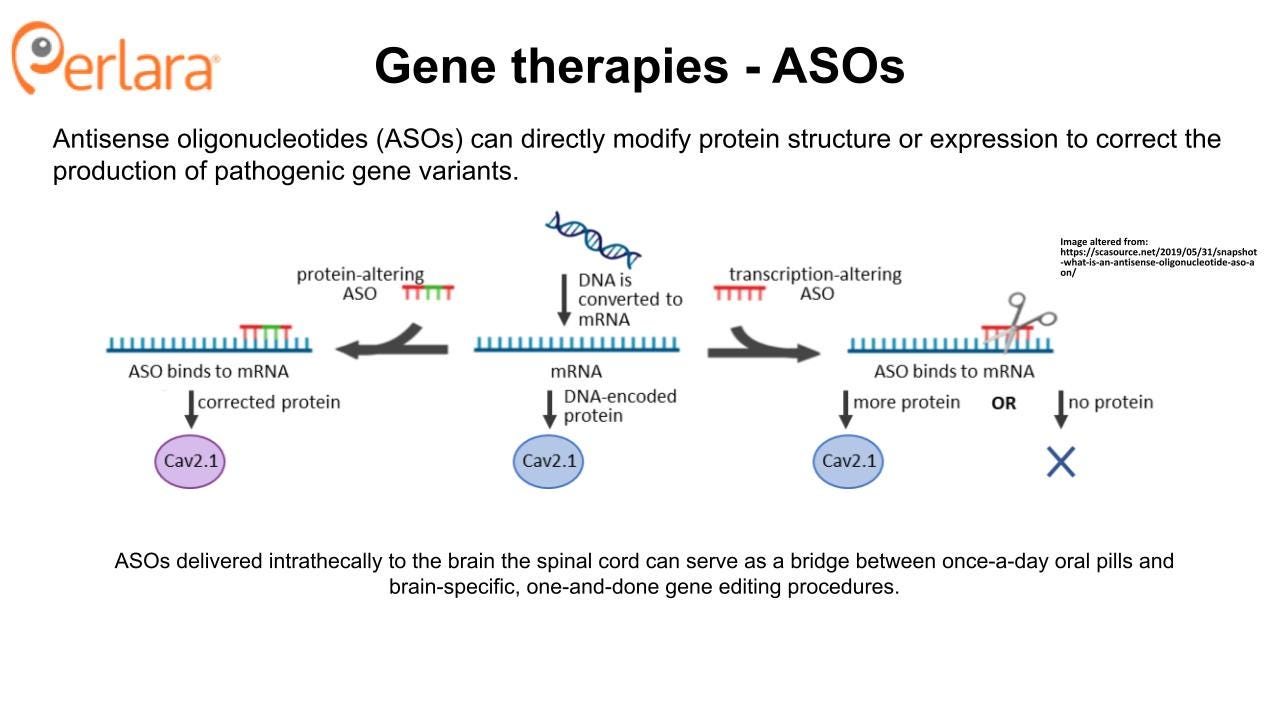
In the next two years, this roadmap proposes that the CACNA1A Foundation establish relationships with recognized scientific leaders in the area of utilizing prime editing for CNS disorders and leverage those relationships to design a multi-stage gene therapy research plan. The CACNA1A Foundation should designate research grant funding toward proposals that aim to address the most critical scientific hurdles to realizing the clinical promise of this technology for CACNA1A-related disorders. At the same time, the CACNA1A Foundation should earmark grant funding to support the development of ASOs that have the potential to work across different LOF or GOF variants versus individualized ASOs for specific named patients with private mutations.
CLINICAL TRIAL AND BIOTECH READINESS
It is still early days, but it is never too early to start forming strategic partnerships with industry and investors who understand that rare genetic neurological diseases are proving grounds for expansion into common neurological diseases and diseases of aging. In this section, we will provide a list of potential partners or individuals that can make introductions to potential partners, and so on. In contrast to other foundations at a comparable point in their development, the CACNA1A Foundation has achieved a precocious level of clinical trial readiness thanks to the natural history study under the supervision of Dr Wendy Chung at Columbia University, which will be a transformative resource when experimental treatments are ready for single-patient clinical studies in the next 6 to 24 months, and thanks to the data sharing partnership with RARE-X, which will help build a vibrant CACNA1A online portal that will stimulate more collaboration interest and partnership interest.
CACNA1A variant data collected from the natural history study will inform the selection of validated disease-related endpoints and clinical biomarkers to establish treatment efficacy and, in some cases, a natural history study can also be used as an adequate control group, or synthetic placebo. The natural history study is the most definitive approach that can be taken at this stage to prepare for small N (3-5) investigator-initiated “pioneer” studies, but much more work will be required in order to establish protocols for a pivotal registration study. The specifics of the CACNA1A Foundation’s future approaches will be dictated by the particular therapeutic modality being pursued in a future clinical trial and pending the results of the ongoing and future research activities enumerated in the Introduction.
We offer the following clinical trial considerations as the drug repurposing programs described in the preceding sections mature and experimental therapies approach the clinic. Roscovitine (as discussed above) and verapamil are the most actionable drug repurposing opportunities on the landscape today for patients with LOF variants and GOF variants, respectively. Promising biomarkers and outcomes measures emerging from the natural history study can be pressure tested in investigator-initiated single-patient IND studies of roscovitine (assuming it demonstrates efficacy in mice) and verapamil. Discovery and validation of rapid-onset and quantifiable EEG, TMS and MRI biomarkers as part of the natural history study could be a game changer, as would wearables (versus daily logs) for monitoring ataxia or seizure activity.
Utilization of repurposed drugs (or individualized ASOs developed by n-Lorem or labs at specialized academic medical centers) in multiple single-patient observational pioneer studies is the optimal way to determine through direct interaction with FDA in a pre-IND meeting whether EEG, MRI or wearables biomarkers are appropriate primary endpoints in a pivotal Phase 3 study. Additionally, Dr. Chung’s natural history study and the work that will be performed by Drs. Poduri and Helbig at CHOP will shed light on a range of neurological symptoms in CACNA1A patients. These results can provide predictive biomarkers for the observational studies based on treatment regimens. Neurofilament light chain (NfL) needs to be explored as a surrogate endpoint especially in severe cases. Seizure reduction is, of course, a validated outcome measure.
The Ataxia Telangiectasia Children’s Project (ATCP) is setting an example for the CACNA1A Foundation to follow to the clinic. We can make the introduction to their leadership team, including the founder Brad Margus, to better understand their go-to-clinic strategy across multiple therapeutic modalities including repurposed drugs and an individualized antisense oligonucleotide/ASO medicine, e.g., atipeksen.
We cannot stress enough that biotech partnership opportunities should be pursued at every stage of research. We recommend cost-sharing initiatives with companies to develop research technology and/or execute pilot studies, which provides access to crucial resources at lower costs, usually in exchange for making the product available to be used by others or the data available for use in publication or promotion. This is a win-win for the CACNA1A Foundation because it both reduces cost and reduces barriers for other scientists to access the same technology or data for their own CACNA1A-relevant discoveries. Similar types of relationships will be pursued for any and all experiments conducted by for-profit companies.
In addition to partnering for research, biotech and pharmaceutical company partnerships will be pursued for clinical development of promising therapeutics that emerge from the robust translational pipeline envisioned by this roadmap. Data establishing the efficacy of a drug candidate in preclinical models will be augmented by learnings from the genotype/phenotype studies which will help determine the target patient population for a given drug and identify relevant biomarkers and clinical efficacy endpoints. Together, these will form an attractive data package to recruit industry partnerships and attract investment, as demonstrated by the Foundation for Angelman Therapeutics/FAST which was able to level up its natural history study by securing an anchor commercial sponsor in Roche.
During the final drafting stages of this document, Perlara brokered an introductory meeting with Octant Bio in order to assess whether their small molecule discovery platform technology was a good fit for CACNA1A. That exchange reinforced the need to develop robust high-throughput cell-based Cav2.1 functional assays, which is currently a rate-limiting step both for deep mutational scanning and drug screening. We also confirmed that the CACNA1A Foundation’s iPSC biobank and natural history study are viewed as essentials for any early-stage platform company like Octant Bio that is considering making CACNA1A a lead program. However, it was acknowledged that precision clinical trials that enroll genetically defined patients with specific variants will be required. The example of Kalydeco was cited as a first-in-class novel small molecule medicine that was initially only approved for use in 4-5% of all cystic fibrosis patients with a G551D variant; patients with other CFTR variants were gradually added to the Kalydeco label as cell-based in vitro functional assays demonstrated drug responsiveness of variants that reduce CFTR function in the same manner as G551D.
Perlara will broker introductory meetings with the following companies and investors with experience and interest in rare neurological diseases to get critical feedback on this roadmap and explore ways of creating public-private partnerships to fund capital-intensive efforts like clinical biomarkers and outcomes discovery and validation, preclinical gene editing in vivo proof-of-concept experiments, and IND enabling toxicology studies that will complement assay development, deep mutational scanning and drug repurposing:
ARCH Ventures – a biotech VC that has backed leading neuroscience companies like Sage Therapeutics, Denali, and most recently Neumora.
Xontogeny – a life sciences accelerator and early-stage fund that specializes in rare genetic diseases.
BridgeBio – a public biotech company with a diversified portfolio of precision medicines targeting rare genetic diseases.
Ultragenyx – a public biotech company with a diversified portfolio of precision medicines targeting rare and ultra-rare genetic diseases.
Beam Therapeutics – a public biotech company with the leading base editing platform technology spun out of Professor David Liu’s lab at Harvard University.
Prime Medicine – a private ARCH Ventures-backed biotech company with the leading prime editing platform technology spun out of Professor David Liu’s lab at Harvard University
As mentioned above, patients with CACNA1A-related disorders exhibit overlapping clinical presentations with disorders that have not been linked directly with CACNA1A mutations, some of which may also benefit from drugs that target Cav2.1. For example, other types of epilepsies have been reported to exist on a spectrum with CACNA1A-related epilepsy (Lennox-Gastaut) or have been linked to CACNA1A in animal models (Childhood Absence Epilepsy). These types of links can mean that drugs discovered to have therapeutic potential for CACNA1A-related disorders may have broader utility as well and may increase biotech and pharmaceutical company interest. The Foundation should also strongly consider applying for a ICD-10 code for CACNA1A-related disorders, which has been successfully done for other neurological diseases like Angelman and Syngap1.
Efficient data sharing will encourage and enable more research into CACNA1A-related disorders and faster progress on therapeutics development. The natural history study led by Dr. Chung and the RARE-X collaboration both have built-in mechanisms for safe (de-identified) and efficient data sharing. Working with Professor Dennis Lal and the Broad Institute to create a CACNA1A online portal for data sharing will provide an additional mechanism that can also incorporate the wealth of therapeutics-enabling data that is available and continues to be generated. The goal will be for all data generated with support from the CACNA1A Foundation to be available via this centralized but open platform.
ROADMAP UPDATES
This Roadmap is considered version 1 (v1) and it will require annual comprehensive reviews to incorporate progress reports and course corrections.