DHDDS Cure Odyssey
Two pioneer families created an Anglo-American alliance and pooled resources in order to fund a yeast-powered drug repurposing project for DHDDS deficiency, an ultra-rare inherited metabolic disease.



In collaboration with



 / Twitter")
Two families, two continents, two medical systems — one, the United Kingdom’s presumably universal National Health Service, the other, the United States’s anything but universal network of public and private payers. Nonetheless the answer the two families, desperate to find what was causing their children’s symptoms, got was the same:
We have nothing, sorry. Pharma doesn’t care about your disease so you’re on your own. We may have more information in 5-10 years. In the meantime, find funding for research. Good luck with that.
And so they did, like so many pioneer families are compelled to do. Cure DHDDS Foundation was set up earlier this year in the UK by Mel and Charles Dixon, whose two children — 14 year old Tom and 8 year old Rosie, pictured below — were found to have variants in the gene, dehydrodolichyl diphosphate synthase, abbreviated DHDDS, which affects protein glycosylation. The Dixons were profiled by ABC News over the summer, making them a rare breed of family that receives earned media.

Joining the Dixons from the US are John Gurley and Zoe Manville aka the band Portugal. The Man, no strangers to the spotlight. Their 12-year-old daughter Frances also has a heterozygous variant — in her case, p.Arg205Gln — in one copy of her DHDDS gene.

To date, only 70 people in the world have been diagnosed with DHDDS deficiency. Internet searches and cold calling scientists, clinicians, anyone who had published anything on DHDDS, brought the two families together and ultimately to Perlara.
These two families have been on a parallel journey, which will be eerily familiar to anyone who has dealt with an ultra-rare disease. The Dixon’s then four year old son, who had developed a tremor, received various diagnoses, from ataxia to autism, before whole genome sequencing identified his heterozygous pathogenic DHDDS variant affecting a different arginine residue, p.Arg37His. (An earlier more selective genetic panel had come back negative).
Frances began exhibiting symptoms during lockdown, when she was nine, myoclonic seizures that cause brief muscle movement and fluttering, that Zoe and John might not even have noticed had they all not been home together 24/7. After months of waiting just to get appointments because of the pandemic, they too received the devastating news that Frances has a rare genetic disease, about which little is known and for which there is no therapy. Both families have been juggling various over-the-counter regimens to alleviate some of the more pernicious symptoms, with no idea on the likely course of the disease, and what the future holds for these children.
DHDDS joins a large list of genes that affect glycosylation. There are well over a hundred such inherited metabolic diseases, collectively known as congenital disorders of glycosylation (CDGs) that have a wide range of health problems from strokes and seizures to development and intellectual delays. DHDDS is not Perlara’s first rodeo dealing with this complicated and essential set of pathways; Perlara is shepherding a dozen CDGs families through drug repurposing protocols, a number of which have advanced to various stages of 1-to-N clinical testing, including a fully enrolled pivotal Phase 3 study with 39 participants.
Tom, Rosie and Frances on the surface appear more mild compared to some in the group with the worst manifestations of mutations in DHDDS, which can include ataxia, seizures, severe cognitive impairment, and even premature death. According to her mom, Frances has had seizures and ataxia, though “they are not intractable yet.” Zoe clarified in an email recently: “I suppose they were spared the severe things early on that some endure.”
Being new on the scene — the first DHDDS variant was described in the literature in 2011 — not much is known about the mechanism or how individual variants produce particular symptoms and phenotypes. No obvious connection between genotype and phenotype has yet to emerge. But it’s early days.
What is known about DHDDS is that it encodes the catalytic subunit of the enzyme cis-prenyltransferase (cis-PTase) which performs the rate limiting step in the biosynthesis of dolichol, a glycosyl carrier lipid (depicted as the yellow rectangle below) required for the biosynthesis of several classes of glycoproteins and glycolipids.
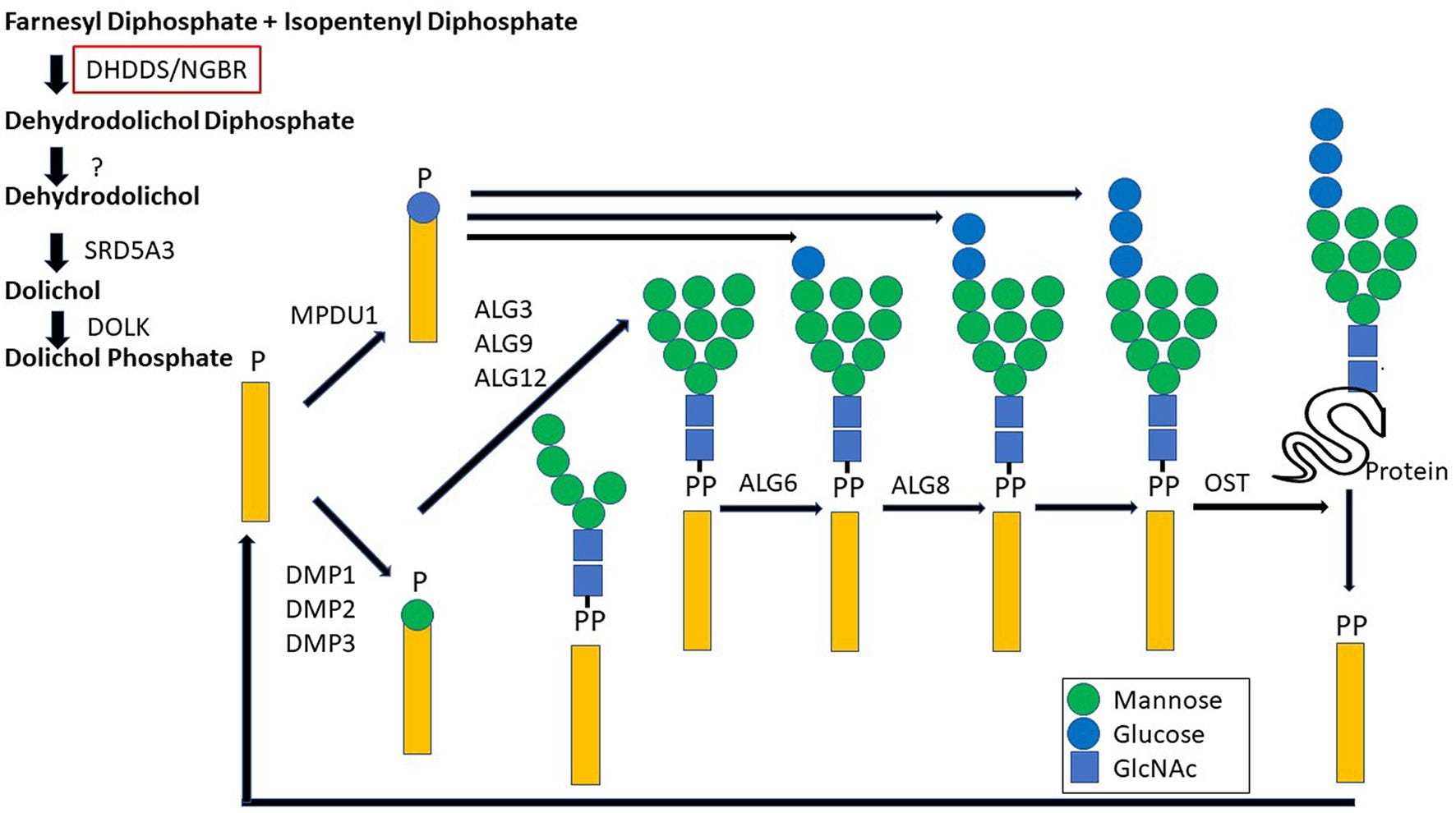
The clinical presentation caused by DHDDS variants are not your typical CDGs, however. For one thing, under or hypo-glycosylated proteins, a hallmark of CDGs, have not been detected in the serum of people with DHDDS variants. Rather, changes in lipid profiles have been seen, which puts DHDDS somewhere between a CDG and a lysosomal storage disease, another class of inherited metabolic diseases with rather severe neurological consequences.
With now 70 known cases of DHDDS variants worldwide, 24 different variants have been reported, almost all de novo mutations. In just a couple of instances, one parent must be a mosaic, which means that only some of their cells have the mutation, among them necessarily germ cells, since the same variant showed up in more than one of their children.
There are three common variants and a bunch more found in only one or two patients; in a detailed study of 25 patients, 19 of them had one of these three common variants — p.Arg211Gln, p.Arg37His or p.Arg37Cys. This same study mapped out which symptoms have been observed with different variants, and some trends are beginning to emerge: one of the more common variants, for example, was associated with a more severe phenotype. Zooming in on Arg37His, Galosi et al., 2022 describes the scene:
In addition, the side chain of Arg37 is stabilized by a network of salt bridges with the β-phosphate group of FPP, Glu89 on DHDDS and the main chain carboxylate group of Lys293 on the C-terminus of NgBR. Introducing a histidine residue at this position would abolish binding interactions with the β-phosphate group of FPP substrate as well as with the C terminus of NgBR.

Although the FPP (farnesyl pyrophosphate) substrate is not visible in this 3-D video rendering by AlphaFold, you can see the salt bridge connecting arginine 37 and glutamate 89. (Arginine 37 starts out behind the play button).
Shifting camera angles for the close-up shot of arginine 205, we return to Galosi et al., 2022 for the intramolecular play-by-play:
Similarly, the side chains of Arg205 and Arg211 form salt bridges with the pyrophosphate of IPP, and replacement of either residue with Gln would eliminate binding interactions with IPP. Although substituting Ser213 with Asn can still maintain hydrogen bonding with the β-phosphate group of IPP, introducing a bulky side chain at this region would result in steric clashes with nearby residues, hence leading to structural destabilization.

Again, although the IPP (isopentenyl pyrophosphate) substrate is not visible in this 3-D video rendering by AlphaFold, you can clearly see that arginine 205 forms a nexus of critical salt bridges without which the enzyme’s active site doesn’t work. The guanidino group of arginine 205 forms a blue trident that starts out pointing to the upper left corner of the screen.
There are several hopeful signs that answers for these and other families with DHDDS children may be forthcoming. The gene is highly conserved evolutionarily, so animal models could provide helpful clues on the mechanisms involved in the pathology and potential targets for therapies. Already mouse models exist that providentially contain these two families’ variants. There is even a zebrafish model for a particular DHDDS variant that uniquely affects the retina. Human fibroblast lines derived from patient samples have been established and more recently several research groups are started on making induced pluripotent stem cells (iPSCs) from the fibroblasts of these children. These cells, which can be differentiated into any cell type with the right kind of coaxing, will provide a research tool for studying the actual cell types affected in this syndrome.
An international team of researchers has been studying the enzyme in a yeast (Saccharomyces cerevisiae) model, in which the natural homolog of cis-PTase (ddol-PP synthase) has been replaced with a number of human variants. Working with these yeast avatars, they found that the enzyme complex formed with the human variants has much less activity both in vitro and in vivo, in some cases, restricting or inhibiting growth to a standstill.
The collection of yeast strains with human DHDDS variants was generously shared with Perlara at the end of last summer. Shout out in particular to Dr Kariona Granbińksa at Yale University. As shown in the figure from her 2022 paper, both R37H and R205Q variants have nearly undetectable cis-PTase activity. The question we all wanted to answer next: could we find compounds that bring these DHDDS yeast avatars back from the brink?

After multiple rounds of seeding density experiments with multiple DHDDS yeast avatars, it turned out that the R205Q variant behaved the best in our hands. Consistent with having < 1% of normal DHDDS enzymatic activity — kissing the lower limit of detection — the R205Q yeast avatar is really sick. That did, however, create a situation where the difference between the mutant yeast and the healthy yeast is huge, so there we’d have no trouble discerning even a modicum of growth rescue.
Z’ optimization experiments were performed twice, once at BAL and again at HTSF, just to be sure. Is the R205Q yeast avatar too sick to rescue? If the accumulation of farnesyl pyrophosphate is toxic to cells, it will take a metabolic miracle. If on the other hand the cells are merely in a quiescent state due to one or more metabolite imbalances, then a small molecule rescuer has a fighting chance to increase growth above the rock-bottom baseline.

We completed the 8,400-compound TargetMol screen the week after Thanksgiving. Much to our excitement, the screen revealed several dozen weak-to-moderate rescuers. None of the rescuers fully rescued the R205Q yeast avatar to wildtype growth levels but that was an impossibly heavy lift. Many of the top rescuers are dietary supplements aka nutraceuticals — ripe for combo therapies to boot.

In the meantime, families have been flocking to the Cure DHDDS Facebook group. Just in the time since Zoe and Mel joined forces, the group has grown from a mere handful to 85 members as of this writing.
Zoe and John are using their platform as world renown rock musicians, to bring attention to their story, which they hasten to add, is the story of millions of other families with children suffering from rare genetic diseases. Frances, a natural performer, has been a part of some of these events.
A recent appearance of John and Zoe on NBC’s Today Show ended with the audio of Frances’s sweet voice, singing her favorite line from one of her parents’ song, “Everything is going to be alright.” Let’s hope so, Frances.