ECHS1 Cure Odyssey
Fibroblasts and flies joined forces in a multi-species drug repurposing campaign that yielded p38 MAPK inhibitors as top rescuers for this ultra-rare mitochondrial disease.


In collaboration with





Disclaimer
The results of the ECHS1 drug repurposing project that we are sharing in the spirit of open science below are novel preclinical research findings and therefore they do not constitute the practice of medicine. Please consult a physician or clinical care team if considering off-label use of any approved drug or compassionate use of any experimental drug. The same caution applies to nutraceuticals, supplements and “generally recognized as safe” compounds.
Three years ago, we embarked on a drug repurposing expedition on behalf of a family affected by ECHS1 deficiency, or ECHS1D, an extremely rare autosomal recessive mitochondrial disease (Leigh-like Syndrome) that typically affects children at birth or infancy. Fly and fibroblast patient avatars were indispensable compasses as we navigated an uncharted landscape, converging on a common disease-modifying mechanism.
ECHS1, which stands for Enoyl-CoA Hydratase, Short Chain 1, encodes an evolutionarily conserved enzyme that localizes to the mitochondrial matrix and is involved in branched chain amino acid (e.g., valine) and mitochondrial fatty acid beta-oxidation pathways that are essential for life (Peters et al., 2014; Ferdinandusse et al., 2015).
Common clinical manifestations include encephalopathy, developmental delay, movement disorders, and cardiomyopathy. To date, there have been less than 100 people — mostly children — identified with ECHS1D around the world (Muntean et al., 2022). Like all ultra-rare diseases, ECHS1D suffers from near-zero awareness and visibility, and little reward incentive for investigation by pharmaceutical companies or academic researchers.
Time and time again, pioneer families fill the vacuum. We made disease modeling forays in worms and flies, scientific firsts. We ultimately opted to focus resources on ECHS1D patient fibroblasts as the primary workhorse for drug repurposing screens, which were performed by Charles River Laboratories. ~10,000 compounds were screened across the Prestwick, Selleck and SPECS libraries. Two metabolic challenge assays were deployed in 384-well plates, at two concentrations (1µM and 10µM), in duplicate. A HTS tour d’force.
Fibroblast hits were funneled into a fly model of ECHS1D for in vivo hit validation studies conducted by Dr. Clement Chow’s lab at the University of Utah. As will be recounted below, those distributed and persistent efforts led to the novel and unexpected discovery of p38 MAPK inhibitors as drug repurposing candidates for ECHS1D. There are currently no approved medicines for ECHS1D.
Perlara’s drug repurposing playbook for inherited metabolic diseases like ECHS1D calls for a portfolio approach and multi-model authentication, i.e., discover compound in model 1, validate compound in model 2. Within a few months of the project launching in the spring of 2021, we booted up three parallel work streams for worms, flies, and fibroblasts.
Evolution dealt a favorable hand for ECHS1D disease modeling, at least on paper. Ancient versions aka orthologs of the ECHS1 gene are encoded in the genomes of animals, including the invertebrates C. elegans (worms) and Drosophila melanogaster (flies):

The worm actually encodes a second ortholog of ECHS1 called ECH-7 in addition to the bona fide ortholog ECH-6. The fly version of the ECHS1 gene has still not been christened in a formal publication, hence the nondescript alphanumeric identifier CG6543. ECHS1D patient-derived fibroblasts are the third model in the mix, in part as a hedge in case the worm and fly models of ECHS1D hit a wall.
Said ECHS1 patient is compound heterozygous for two different missense variants: N59S and T124I. N59 is evolutionarily conserved in all animals. While T124 is a more variable position, the hydrophobic amino acid isoleucine is not observed at this site in the animal kingdom. The N59S and T124I variants are computationally predicted to be damaging – N59S potentially more so – but the exact loss-of-function defect caused by each mutation has not yet been elucidated.
InVivo Biosystems (IVB), experts in CRISPR-edited animals for biomedical discovery, was commissioned to create ECHS1 knockout worm avatars — an ech-6 single knockout mutant and an ech-7 single knockout mutant — as well as a worm avatar expressing the N59S variant in the ECH-6 gene, highlighted in green below.

According to Wormbase, RNAi-mediated knockdown of ECH-6 produces a variety of phenotypes from slow larval growth to defects in foraging behavior. However, a ech-6 mutant expressing patient-derived variants had not been generated or studied to date by any lab.
IVB conclusively showed that a homozygous ech-6 knockout worm avatar exhibits 100% larval lethality, while the homozygous ech-7 knockout worm avatar is viable but developmentally delayed. We reasoned that the fully penetrant larval lethal phenotype of the ech-6 knockout would be difficult if not impossible for a compound to rescue in the context of a drug repurposing screen. That was our first dead-end.

Disappointingly and somewhat curiously, the ech-6 N59S worm avatar did not exhibit lethality or statistically significant developmental delay (blue-filled circles). The sticking point with the worm model of ECHS1D was the fact that expression of the human ECHS1 gene in the ech-6 knockout worm avatar did not rescue lethality. What’s more, the ech-6 N59S worm avatar behaved unexpectedly upon exposure to excess dietary valine, meaning indistinguishable from the wildtype worm strain (N2).

People living with ECHS1D may benefit from valine-restricted diets (Ganetzky et al., 2016), so we hypothesized that the ech-6 N59S worm avatar might also be intolerant of high valine supplementation. Alas, we observed no change in valine sensitivity, as shown in the above figure. In fact, the ech-6 N59S variant engenders resistance to high valine exposure when expressed in the ech-7 knockout genetic background (purple-shaded bars).
We made one last-ditch effort to salvage the ECHS1D worm modeling project and identify a phenotype amenable to rescue in a high-throughput drug repurposing screen. We treated the ech-6 N59S worm avatar with excess propionate, a short-chain fatty acid that is normally metabolized by ECHS1. As shown below, 120mM propionate supplementation did not affect the growth of the ech-6 N59S worm avatar one bit.

All told, the ECHS1D worm modeling exercise consumed six months. No disrespect to worms, which can be perfectly suitable patient avatars when there’s a biological fit. However, the 100% lethality of the ech-6 knockout worm avatar, the risk of moving forward with the ech-7 knockout given the unexplained behavior of the ech-6 N59S, ech-7 knockout double mutant, and the failure to find sensitizing environmental conditions made a high-throughput primary drug screen with worms too risky. We swiped left on worms and focused our attention on flies and fibroblasts.
Soon after the InVivo Biosystems project was in motion, we commissioned a biotech startup called Vivan Therapeutics to create an ECHS1 RNAi knockdown model in flies. The attempt with flies proved successful: ECHS1 knockdown is larval lethal, consistent with the larval lethality of the ECHS1/ech-6 knockout worm.
In contrast to the worm, knockdown of ECHS1 expression is tunable by temperature in flies using a nifty array of genetic tricks.

At the standard growth temperature (18˚C/64.4˚F), the GAL80 protein sops up the GAL4 protein, inhibiting its function. When flies are reared at a balmy temperature (29˚C/84˚F) the GAL80 protein wilts from the heat, freeing the GAL4 protein to activate expression of a gene-silencing cassette that specifically shuts off ECHS1 expression in all fly tissues. The activity of GAL80, and by extension the level of ECHS1 knockdown, is inversely proportional to the temperature.
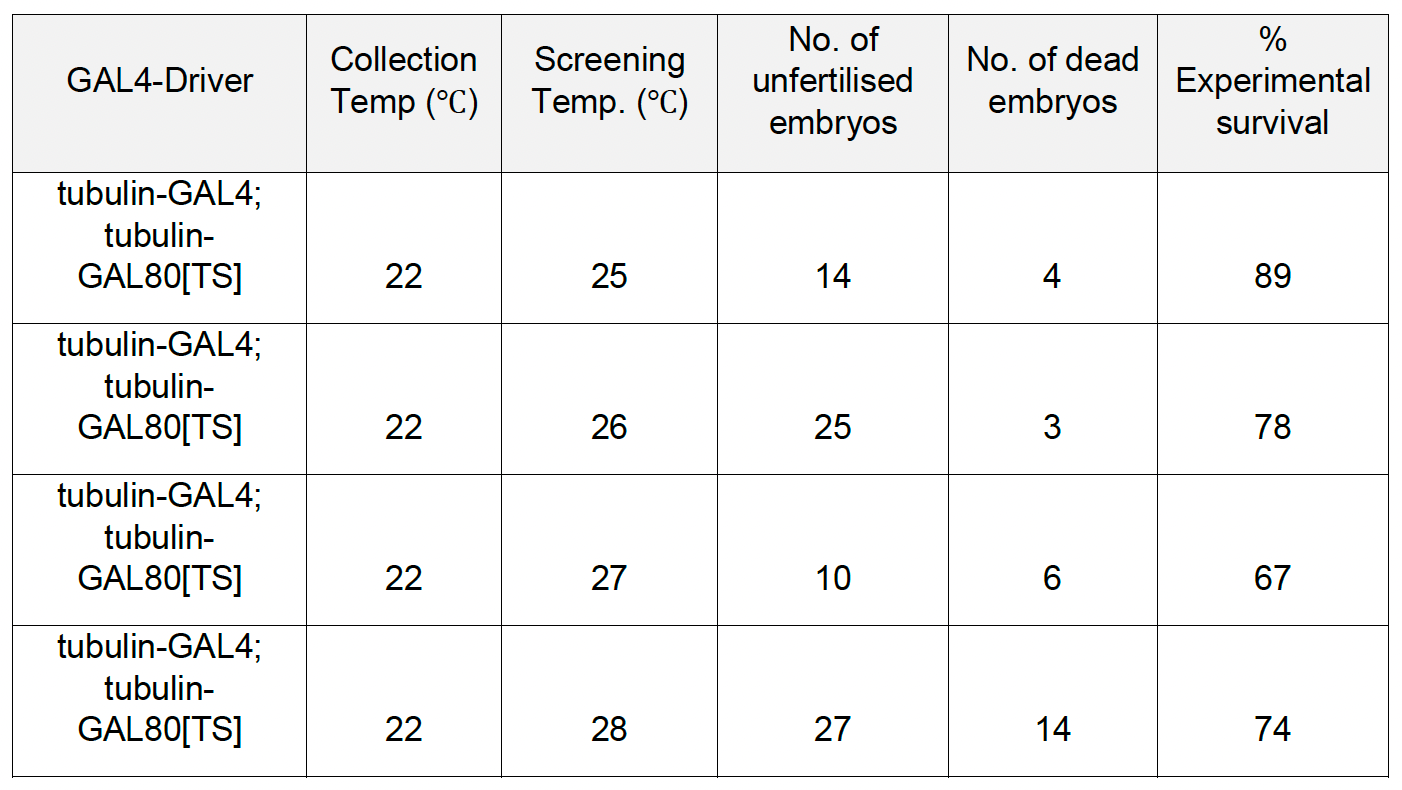
Results of a temperature scan experiment to determine the optimal level of lethality are summarized in the table below.

Notice that at 28˚C, there were surviving larvae or pupae. The Goldilocks temperature is wedged between 26˚C and 25˚C. Importantly, the timing of lethality was shown to be during larval stages, not at the embryonic stage. If lethality emerges too early in development it would not be possible to rescue in a drug repurposing screen because larvae have to be viable enough to feed and ingest the test compound dissolved in the fly food.
We faced a fork in the road: fibroblast or fly. As with the worm model of ECHS1D, we were concerned at that time that larval lethality might be too difficult to overcome in a drug repurposing screen. So we held the ECHS1 fly model in reserve for secondary hit validation studies and proceeded with a primary screen in fibroblasts.
In the Fall of 2021, CRL was expanding the ECHS1D patient fibroblast line alongside a healthy age-matched and sex-matched control fibroblast line. Based on the successful execution of a previous drug repurposing project conducted at CRL using a different mitochondrial disease patient-derived fibroblast line, we were reasonably confident that ECHS1 patient fibroblasts could serve as the primary drug screening disease model.
We challenged ECHS1 patient fibroblasts with growth in two different stress conditions, or metabolic challenges: galactose supplemented media and propionate supplemented media.

We knew from a previous drug repurposing project that mitochondrial disease patient-derived fibroblasts grow normally in media supplemented with glucose as the sole carbon source, but they grow poorly in media supplemented with galactose as the sole carbon source. Galactose forces mitochondrial disease patient-derived fibroblasts to utilize their mitochondria to fuel cell growth and division.
Thinking about other ways to metabolically challenge cells, we hypothesized that ECHS1D patient-derived fibroblasts would exhibit hypersensitivity to excess exogenous propionate, which increases the levels of toxic ECHS1 substrates. In other words, ECHS1 patient fibroblasts “fail to thrive” in media supplemented with either galactose or propionate.
CRL performed assay optimization experiments in galactose and propionate challenge media separately because we decided to pursue a two-track drug repurposing screen: a galactose challenge assay and a propionate challenge assay. We reasoned that compounds that rescue both ECHS1 patient fibroblasts in both challenge paradigms would be more likely to have therapeutic potential because they might alleviate both the bioenergetic defect observed in ECHS1 patients (galactose challenge) as well as the buildup of toxic metabolites upstream of ECHS1 (propionate challenge) observed in patient fibroblasts and in patient biosamples such as urine or blood.

In the beginning of 2022 — nine months after we initially set sail — CRL performed dual pilot screens using a small collection of ~2,500 bioactive compounds (the Prestwick and Selleck libraries), followed by a more exhaustive screen of the ~7,500-compound SPECS (aka Broad Repurposing Hub) library. In both instances, test compounds were screened at 1μM and 10μM so that we could stratify hits by potency.
Over 100 hit compounds from the galactose challenge screen and the propionate challenge screen were reordered, solubilized powder stocks as fresh solutions, and then tested in dose-response assays in both challenge paradigms. (We never subjected ECHS1 patient fibroblasts to simultaneous galactose and propionate challenges).

Hit validation studies confirmed p38 MAPK inhibitors, e.g., losmapimod, as the top cluster. p38 MAPK inhibitors were among the only class of primary screening hits that validated in both the galactose challenge condition and the propionate challenge condition. Here’s the potency determination results for losmapimod in galactose challenge.

Next, we evaluated the p38 MAPK inhibitors and several other mechanistically unrelated hit compounds in a fly ECHS1 RNAi knockdown model. It was one thing to observe modest enhancement of growth of fibroblasts by rescue hits in response to two physiologically relevant metabolic stresses. It’s literally another beast entirely to observe rescue effects of a test compound in a whole animal.
The Chow lab created an ECHS1 RNAi knockdown line for hit validation experiments. Just as Vivan Therapeutics observed with their temperature-sensitive ECHS1 RNAi knockdown line, the Chow lab showed that ECHS1 knockdown larvae are developmentally delayed and drastically smaller in size compared to the control.

A summary of the effect of four p38 MAPK inhibitors — losmapimod, talmapimod, ralimetinib and neflamapimod — on larval size is shown below. The top row labeled ECHS1 RNAi is the negative control: we expect no growth in the absence of intervention. Statistically significant rescue of the larval size defect was observed for 25µM losmapimod and for 25µM and 50µM talmapimod.

Remarkably, all of the p38 MAPK inhibitors rescued not only larval developmental delay, i.e., drug-treated larvae were bigger than untreated control larvae, but they also rescued the “sinking” phenotype. Normally, wildtype larvae sink to the bottom of a well if floated on top of a well filled with liquid. ECHS1 RNAi knockdown larvae float instead of sinking but ECHS1 RNAi knockdown fly larvae treated with p38 MAPK inhibitors drop like anchors, though not to the degree of the wildtype fly larvae.

Together, these fly hit validation studies demonstrate that p38 MAPK inhibitors rescue ECHS1 deficiency in vitro in patient-derived fibroblasts and in vivo in ECHS1 RNAi knockdown fly larvae. Going from fibroblast to flies led to this discovery. Would we have arrived at the same place if the directionality was flipped?
Meaning, if we had run a fly screen and a fibroblast screen at the same time, would the two models have converged on the same answer? Turns out the answer is, yes! A group of ECHS1 families crowdfunded a fly screen at Vivan Therapeutics after we completed the fibroblast screening campaign. Lo and behold, Vivan identified losmapimod as the top rescue hit of fly larval lethality.
There is compelling support for the use of p38 MAPK inhibitors in mitochondrial diseases like ECHS1 deficiency. An emerging body of literature from Professor Simon Johnson’s lab (Stokes et al., 2022), Professor Peter McGuire’s lab (Kapnick et al., 2018) and others, suggests that Leigh and Leigh-Like Syndrome mitochondrial diseases have an immune cell component to pathophysiology. p38 MAPK inhibitors were originally developed for autoimmune diseases with the immune cells as the primary target tissue. It seems drug repurposing works in not so mysterious ways.
Losmapimod is currently in clinical development by Fulcrum Therapeutics for adults living with another rare disease called facioscapulohumeral muscular dystrophy (FSHD). Plans are underway for expansion into the pediatric FSHD population. In the meantime, we’re planning mouse efficacy studies to determine the range of effective and well-tolerated doses of p38 MAPK inhibitors in a mammal. While we wish the path to the clinic were more straightforward, the science has to take its course. Onward with our search for a cure to ECHS1D!