FOXG1 Cure Odyssey
A mother’s “darkest days” leads to a foundation and a magazine dedicated to families caring for children with rare diseases.



In collaboration with


 / Twitter")
When genetic lightning strikes
Megan Nolan knew early on that something was wrong with her baby, Domenico. When he was just a few months old, she noticed some disturbing behaviors: not making eye contact, atypical limb movements, and hours-long crying spells that could at times go all night.
During a 14-month diagnostic odyssey spanning two continents, Megan and her husband consulted a dozen doctors, none of whom provided any answers or suggested genetic testing.
"Had even one pediatrician mentioned that it could be due to a genetic abnormality, we would have paid for genetic testing ourselves,” Megan Nolan.
When Domenico was 14 months old. Megan and Kamal, who at that point felt like they were “grasping at straws”, moved forward with double eye surgery for Domenico’s strabismus, thinking that it might be the cause of some of his movement and visual problems.
After the surgery, perhaps coincidentally, perhaps not, Domenico had his first seizure. This, combined with his medical history, brought a neurologist at CHOP to recommend testing with a panel of genes associated with epilepsy and seizures caused by a variety of causes. Two months later, they had a diagnosis — FOXG1 syndrome (FS), a rare genetic disorder affecting a master regulator of neurological development.
Finally, they had the answer they had been seeking, but it still left them with a severely disabled child and no idea how to manage an increasing number of symptoms or what the future holds for their child and their family.
FUNCTIONS OF FOXG1

What is FOXG1?
FOXG1 stands for fork-head box G1, aka brain factor 1, a member of the fork-head box family of transcription factors. Transcription factors, considered master regulators of gene expression since they control the expression of groups of genes. are highly conserved proteins, which affect a number of different organ systems.
In the case of FOXG1, the target organ is the brain. The name, fork-head comes from the first FOX gene to be discovered, which happened to be in fruit flies Drosophila melanogaster); mutations in the fly version of FOX causes gnarled fork-like bristles, whence the name fork-head comes.
FOXG1 switches on early in fetal brain development and is essential for the proper formation of a functional brain cortex, interacting with a large number signaling pathways and processes.

It’s no wonder then that FS patients display serious brain abnormalities, such as microcephaly and disruptions of the corpus callosum, which connects the right and left sides of the brain. The children exhibit numerous symptoms, which vary in severity, including intellectual disability, dyskinesis of movement, absence of verbal and language acquisition, irritability as well as some autistic features.
The first heterozygous de novo mutation in FOXG1 was identified in a 7-year-old child in 2005, who had microcephaly, complete disruption of the corpus callosum, and severe cognitive deficits. As genetic testing has become more commonplace, the numbers of FOXG1 patients has been steadily growing, and may be close to a thousand now.
The FOXG1 gene has 4,890 base pairs, with a single exon (uninterrupted coding region) and multiple binding domains.

Most FS cases are caused by de novo mutations, although there are a few cases in which the parents appear to be mosaics, which means only some of their cells (presumably germ cells, as the trait was passed on to their children) have the mutation.
FOXG1 mutations, of which there are more than 100 (as of January 2023), come in all varieties — missense, frameshift, deletion and even duplications. All result in a state of FOXG1 haploinsufficiency, or the inability of the body to cope with only 50% FOXG1 gene expression.
Mapping studies have shown that mutations can occur anywhere along the gene and in all binding domains, but there are several hotspots, mainly in areas where runs of single bases occur--- c.256dupC and c.460dupG, which have seven repeated cytosines (CCCCCCC) and guanines (GGGGGGG) respectively.
These kinds of sequences are prone to replication errors. Studies connecting genotype to phenotype are inconclusive, but the severity of symptoms decreases as you move from the N-terminus to the C-terminus of the protein.
DISTRIBUTION OF MUTATIONS ALONG THE FOXG1 GENE

The awesome power of yeast genetics
FOXG1 Syndrome (FS) has two things going for it. First, FOXG1 is well-studied as rare disease genes go — over 600 publications on PubMed. Second, FS sits at the upper end of the ultra-rare disease distribution, meaning multiple groups have funded a portfolio of research projects over the years, and new families are unfortunately struck by genetic lightning each year. Although yeast models of FS have been studied with support from the FOXG1 Research Foundation, these results are not published and we are not aware of any ongoing or completed FS drug repurposing projects using yeast.
Yeast models with gene defects often exhibit growth defects. Slow growth can be
rescued in a pathway-agnostic high-throughput drug repurposing screen. The yeast gene HCM1 is the ortholog (equivalent) of human FOXG1. HCM1 performs the function of many FOX family members in humans. This is a fairly typical situation — humans have about five times as many genes as yeast, and many of these new additions arose through gene duplication events over the course of evolutionary history.
Understanding that one can talk themselves out of almost any experiment that hasn’t been tried before, we set out to rescue growth defects of HCM1 mutant yeast. In our experience, in cases where yeast have a single ancestral gene that diverged into an entire family of genes in humans, screening using yeast lacking that ancestral gene has produced biologically-relevant results. This fact, combined with the fact that yeast screens are expeditious and cost-effective, led us to recommend this yeast-driven approach.
We conducted growth assays including both haploid and diploid FOXG1 (yeast HCM1) deletion strains, along with their respective controls, under two temperature conditions: 30°C (ambient) and 37°C (heat shock). We observed a significant growth impairment in the diploid HCM1 knockout strain at 30°C.

After several failed attempts at further optimization, we pivoted away from using high temperature as a sensitizer to using a pharmacological sensitizer. Based on FOXG1 biology in yeast and human cells, we reasoned that HMG1 mutant yeast would be sensitized by the microtubule stabilizer benomyl.
Here’s the initial seeding density experiment using benomyl sensitization to reveal a growth defect.
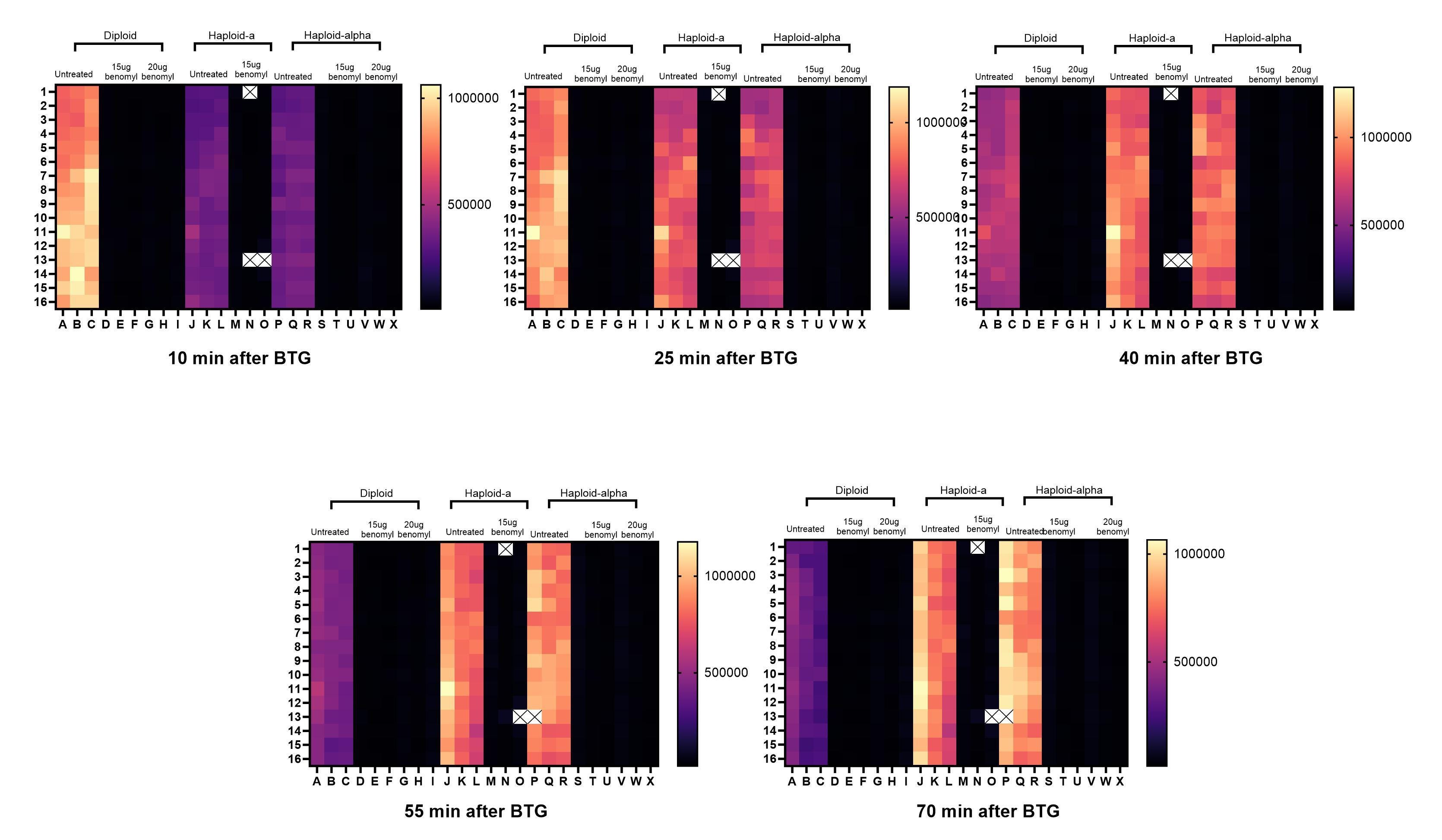
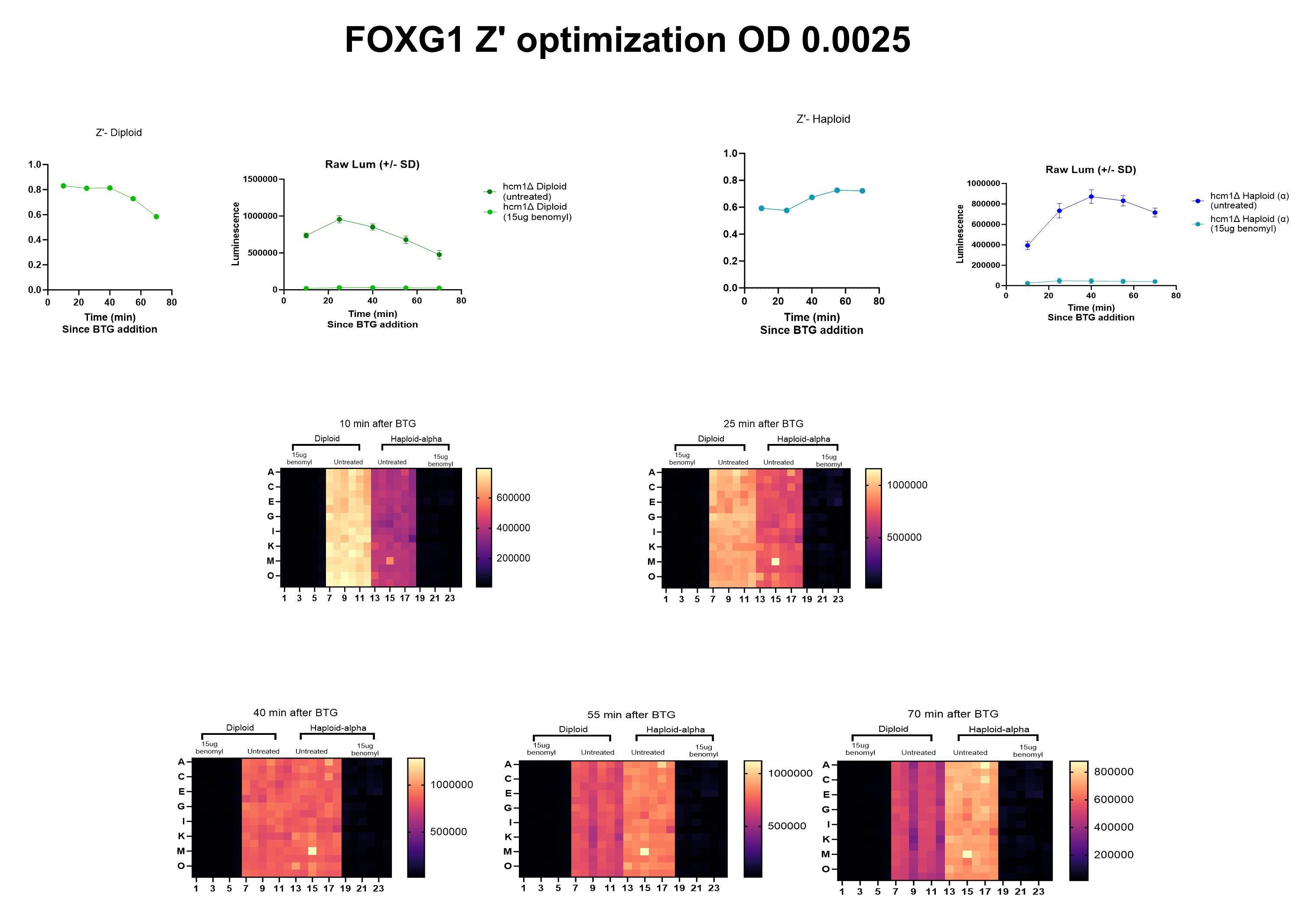
Next, our yeast team completed a dose-finding Z’ optimization experiment at BADASS Labs to determine which concentration of benomyl to advance to the screening stage. That Goldilocks dose turned out to be 15µg/mL.
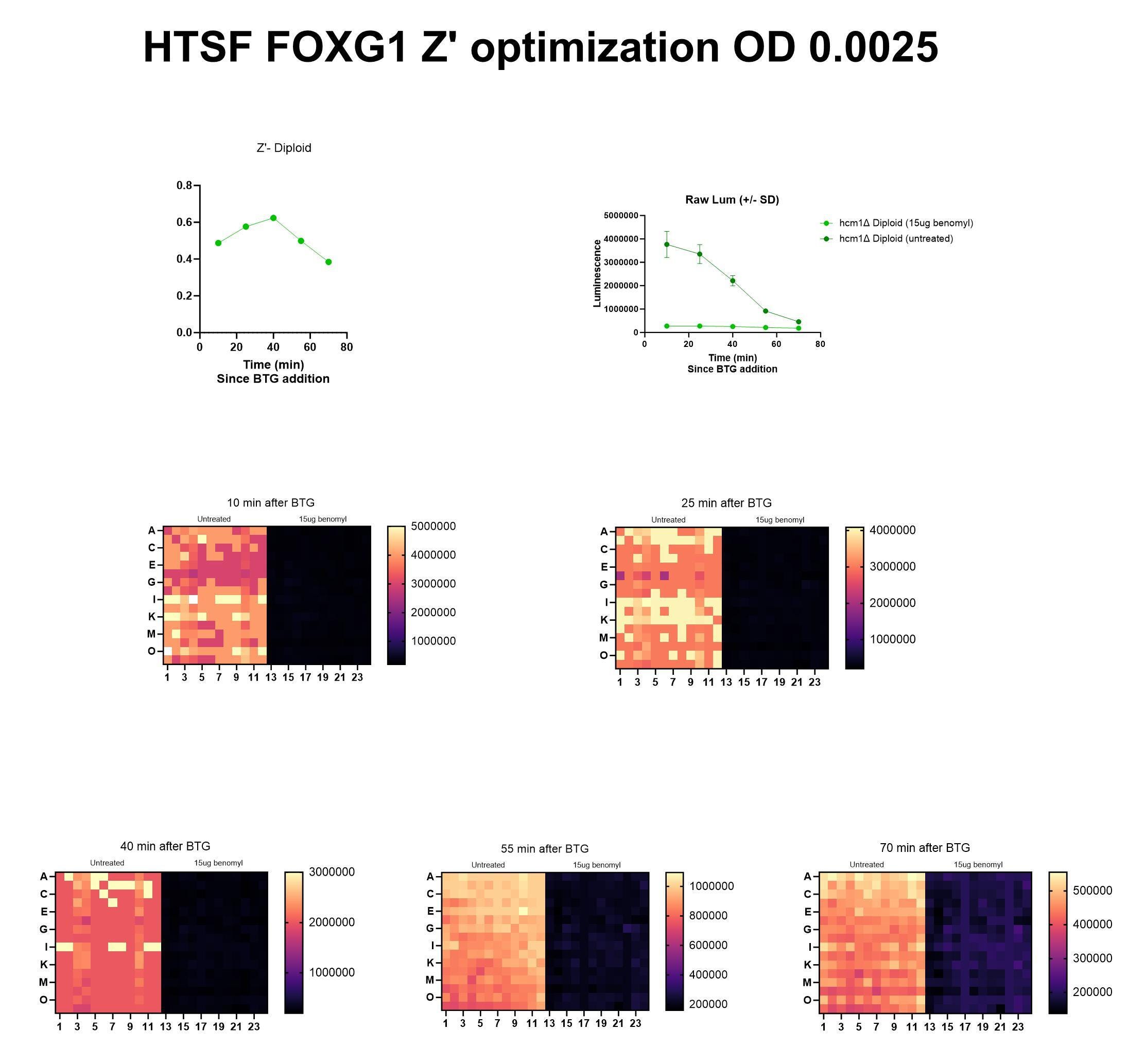
Here’s the Z’ optimization experiment performed at HTSF using the 15µg/mL dose.
The TargetMol screen was just completed today, auspiciously on the eve of Rare Disease Day 2024. More details to come soon.
Rare Uncommon Disease Day
In thinking about all the other families — potentially millions of them — who find themselves in the same situation, as she did, Megan decided to use the skills that she had developed in media and advertising, to fill in these gaps of knowledge and to create support systems, systems that were non-existent when Domenico was diagnosed.

In these situations, parents become care givers overnight. The job change—given a new job title, without a job description---we’re not given any training and we’re not told what our future looks like, often not given any support or support system. Megan Nolan
Thus beget The Children's Rare Disorders Fund (CRDF), an ultra-lean non-profit biotech, founded by Megan and her husband, Kamal Sandu, in July, 2022, just three months after their son was diagnosed. The CRDF’s goal is to support research into therapies for rare diseases. While inspired by Megan and Kamal’s son, the foundation is exploring a variety of modalities that could be applied to any or all underserved rare disease.
Among the modalities is, of course, drug repurposing. Perlara’s work on FOXG1 syndrome is the first project to receive funding from CRDF, which came into existence in July 2022, just three months after Domenico’s diagnosis.
More recently, Megan launched a journal, Rare Parenting, a treasure trove of information for families in need of guidance and information, what Megan calls "service journalism." There you can find articles that provide step-by-step guidance on what parents of undiagnosed children can do, what kind of symptoms might be a sign of neurological disorders, financial planning to mention a few.
To anyone reading this who is interested in drug repurposing screens: the TargetMol library used here was provided by the UCSC Chemical Screening Center, spotted to assay plates using our acoustic dispenser and lab automation. These and other high throughput and cell imaging technologies and screening assays are available here for any customer and we're open for business! Please check out our website if you are interested in collaborating: csc.ucsc.edu/mission. To contact us, email brabbitt@ucsc.edu. We're open for business. Thanks!