HNRNPH2 Cure Roadmap
Our 7th Cure Roadmap was commissioned by To Cure A Rose Foundation based in Austin Texas, which popped up its own lab called Everlum Bio to build iPSC neuron models and advance cure-focused research.


HNRNPH2-related Disorders Cure Roadmap
Prepared for To Cure a Rose Foundation by Perlara PBC
March 2022

Vision
Develop a disease model of the predicted pathogenic p.R114W missense variant in HNRNPH2-related neurodevelopmental disorder (Bain Syndrome) and develop therapeutic strategies to target or correct this specific variant. This multi-year, multi-modality roadmap aims to lay out the scientific and therapeutic strategies to find a treatment for patients with HNPNRH2-related neurodevelopmental disorders (NDD). Based on the current knowledge base around Bain Syndrome and the outcomes of the basic science work we outline, we recommend moving forward with two therapeutic approaches for this condition. Furthermore, this roadmap includes recommendations for the “To Cure a Rose (TCAR)” foundation for prioritization of their work based on the current knowledgebase. Thus, this roadmap will focus primarily on finding a therapy for Rose (Rosie) McPherson, a patient who harbors a HNRNPH2 mutation at p.R114W.
Executive Summary
The goal of the TCAR Cure Roadmap is to stimulate development of medicines that will treat (and potentially cure) HNRNPH2-related NDD.
Our Key recommendations for the next 12-24 months for TCAR are:
Basic Science 1: Develop an in vitro model for HNRNPH2-related NDD by investing in efforts to develop patient-derived iPSCs and further differentiate them into neuronal cell types.
Basic Science 2: Identify the function of HNRNPH2 and its downstream targets by using NGS technologies (genomics and transcriptomics) in an effort to define HNRNPH2 variants.
Basic Science 3: Develop an in vivo model for HNRNPH2-related NDD by collaborating with Jackson Lab to create mouse models carrying humanized versions of HNRNPH2.
Basic Science 4: Reporter assays – Synthesize the results from BS1 and BS2 to build dual color reporters that can be utilized for drug and ASO screens. Additionally, search for other high-throughput screenable phenotypes.
Therapeutic Track 1: ASO design – Design allele-specific and allele-agnostic ASOs to target HNRNPH2 variants and test these in the in vitro and in vivo models.
Therapeutic Track 2: Drug repurposing – Screen for small molecules that alleviate variant-specific phenotypes in cells.
Fundraising and Project Management: Throughout the timeline, work closely with Perlara to identify and improve our fundraising efforts.

Authors
Courtney Banks, PhD (Cure Guide, Perlara PBC)
Arun K. Ramani, PhD (Cure Guide, Perlara PBC)
Ethan O. Perlstein, PhD (CEO of Perlara PBC & Maggie’s Pearl LLC)

Introduction
Among the hundreds of candidate genes proposed for neurodevelopmental disorders (NDDs), genes involved in RNA metabolic processing and regulation of gene expression have been shown to be enriched for de novo variants among probands with NDDs [1]. RNA processing (splicing, transport, localization, translation, and degradation) is critically important for brain development and function, as neurons are post-mitotic cells dependent on RNA expression, as well as spatiotemporal isoform specificity, for individual growth and functionality [2]. To successfully regulate RNA processing and protein synthesis, over 500 RNA-binding proteins (RBPs) in humans are abundantly and ubiquitously expressed, found primarily in the nucleus [3]. Although ubiquitous, there are tissue-specific changes in alternative splicing from RBPs resulting in cell-specific phenotypes [4]. As RBPs are necessary for many steps of neuronal RNA metabolism, there are multiple opportunities for dysfunction, which is highlighted by the range of neurological phenotypes resulting from variation in RBP-encoding genes, including neurodegenerative diseases, muscular atrophies, and various cancers.
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are a subfamily (consisting of 33 core and minor members) of RNA binding proteins that form a complex with heterogeneous nuclear RNA (hnRNA) and are implicated in many steps of RNA processing [5]. These proteins have distinct nucleic acid binding properties, are associated with pre-mRNAs in the nucleus, and influence pre-mRNA processing and other aspects of mRNA metabolism and transport. While all members of the subfamily are present in the nucleus, some also shuttle between the nucleus and the cytoplasm. Heterogeneous nuclear ribonucleoprotein HNRNPH2 is a member of this subfamily and is located on the X-chromosome. It is ubiquitous in expression and is highly colocalized in the nuclear compartments in the brain, gastrointestinal tract, lung, skin, spleen and testes. It is also found localized in the cytoplasm and is suspected to shuttle back and forth. HNRNPH2 harbors three quasi-RNA Recognition Motifs (RRMs) that bind RNA, two glycine-rich domains that aid in homo/heterodimerization, as well as a nuclear localization signal (NLS) that is necessary for its localization to the nuclear compartment.

Variants in the X-linked gene Heterogeneous nuclear ribonucleoprotein HNRNPH2 (HNRNPH2) cause a neurodevelopmental syndrome (a rare disease) identified by Drs. Bain and Chung in 2016 and is a type of X-linked syndromic intellectual disability (classified as Bain type of X-linked syndromic intellectual disability) [5]. Since then, over 100 individuals (male and female) have been identified with a suspected prevalence of 1:50,000–1.5:100,000 live births. Although there is currently no positively defined phenotype, the main characteristics may include: developmental delay/intellectual disability, severe language impairment, motor problems, growth and musculoskeletal disturbances, dysmorphic features, epilepsy, autism spectrum disorder, cortical visual impairment, and rarely, early stroke and premature death [5,6].
Additionally, another member of the subfamily, HNRNPH1 is a highly conserved paralog of HNRNPH2 with over 95% sequence homology at the DNA level. Individuals with missense mutations in HNRNPH1 display overlapping phenotypic characteristics to those carrying mutations in HNRNPH2. Assuming that HNRNPH1 is fully functional in individuals with HNRNPH2-related disorders and that these two proteins function similarly, functional HNRNPH1 may be able to partially compensate for disrupted HNRNPH2. Assessing the functional consequences of each of these de novo variants will be important to better understand the pathophysiology and identify possible therapeutic interventions for these disorders.

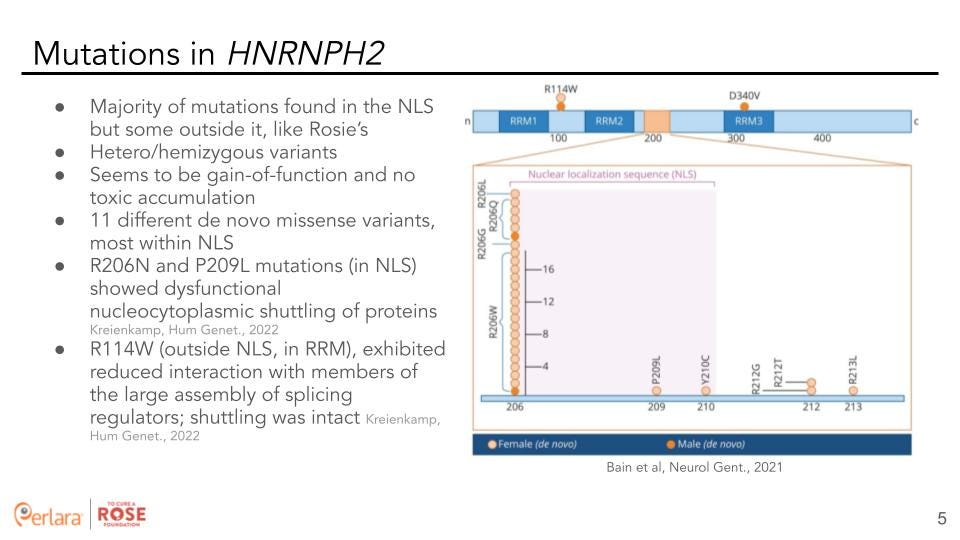
The majority of variants (eleven different de novo missense variants) in HNRNPH2 are localized in the NLS but some are located outside it. All of these are either heterozygous (in females) or hemizygous (in males) variants and seem to be gain-of-function with no toxic accumulation. R206N and P209 L mutations (in NLS) showed dysfunctional nucleocytoplasmic shuttling of proteins. Whereas, the R114W mutation (outside NLS, in RRM) exhibited reduced interaction with members of the large assembly of splicing regulators but shuttling was intact [7].
Currently, no therapeutics exist that restore cognitive or motor function.

Our Proposed Strategy:
Given that there is very little that is clearly understood about HNRNPH2 and its mechanism, we believe that it is necessary to first seek to answer some basic mechanistic and functional questions before we can proceed with therapeutic approaches. Some of the limitations to our present understanding of HNRNPH2 include:
What is the biological role of HNRNPH2?
If we assume that HNRNPH2 predominantly functions through regulating mRNA maturation, what are its downstream targets?
What are the primary targets? Are these specific to certain (neuronal) tissues and developmental time points?
Since HNRNPH2 is X-linked and both males and females are affected, what is the mode of action of HNRNPH2? Is it a loss-of-function or a gain-of-function phenotype?
Are there potential compensatory mechanisms within the HNRNP sub-family? Would HNRNPH1 or other paralogs compensate for HNRNPH2? What is the compensatory mechanism?
We are fortunate that the work done by Drs. Bain, Ricupero, Lessel and others have generated a wealth of knowledge and triggered interest in the study of HNRNPH2 and the biology underlying the related neurodevelopmental disorders. With this information in hand, the To Cure a Rose Cure Roadmap envisions advancing toward new approaches along three main tracks: a) basic research to extend our understanding of HNRNPH2 biology and how mutations alter its biology b) a therapeutic track with antisense oligonucleotide (ASO) strategies, and c) an alternative therapeutic strategy utilizing drug repurposing.
Modalities to Consider
To get us closer to therapeutic readiness, identify targets, and develop medical interventions we require disease models, patient-derived samples, and a deeper understanding of the pathophysiology of HNRNPH2 variants. The majority of mutations in HNRNPH2 are found in the NLS, with the R206N and P209L mutations showing dysfunctional nucleocytoplasmic shuttling of proteins. On the other hand, the R114W mutation that lies outside the NLS (and the causal mutation for Rosie) exhibits reduced interaction with members of the hnRNP complex [7].
An important question to address, therefore, is whether to work specifically with the R114W mutational background, the more prevalent R206N background, or both. Since clinical outcomes associated with these two backgrounds are different [7], the initial focus of this proposal will be on the R114W mutation affecting Rosie. It is to be noted that the results and the understanding gained from this study can be broadly extended to a mutation agnostic approach that could become applicable to HNRNPH2 related NDD at-large.
Interestingly, HNRNPH2 is highly expressed in fibroblasts and Rosie manifests tougher and more sensitive skin. Because fibroblasts would be a much cheaper cell type to perform drug and ASO screening in, it is worth considering trying to find a phenotype for her fibroblasts against control fibroblasts to see if we could find a potential marker for high throughput screening.
It is of note that the strategic alliance team at St. Jude’s (specifically Dr. Paul Taylor’s lab which focuses on HNRNPH2) is formulating a research update to share with HNRNPH2 advocacy groups. The Yellow Brick Road Project group is one of the advocacy groups involved. Although this process might involve signing an NDA and joining the strategic alliance, the research update would be extremely beneficial for understanding the pathophysiology of HNRNPH2 variants and, in turn, making a more informed decision on ASO design.

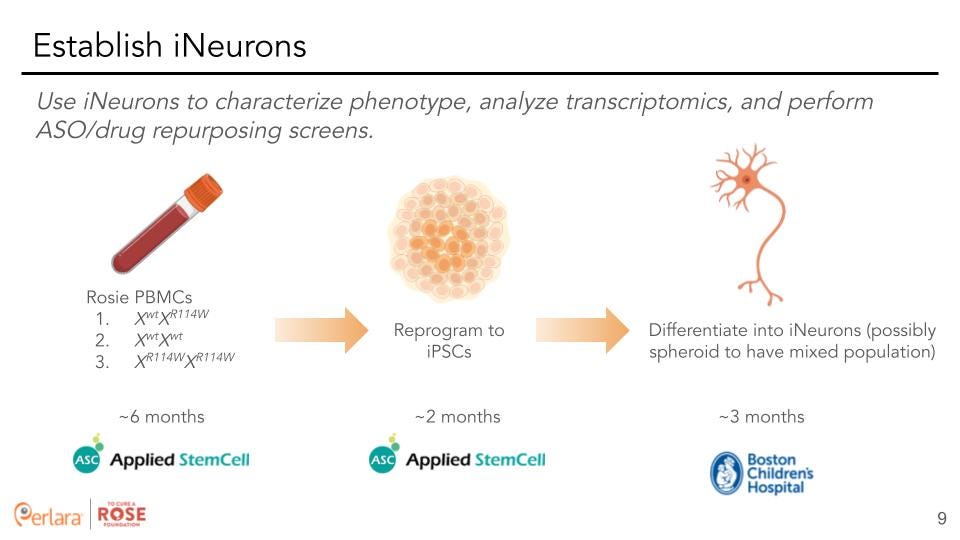
Basic Science 1: Develop an in vitro model for HNRNPH2-related NDD by investing in efforts to develop patient-derived iPSCs and further differentiate them into neuronal cell types.
Stem cell disease modeling using iPSCs is a powerful approach that opens up opportunities to investigate unique cellular phenotypes and uncover pathogenic mechanisms. It also allows us to probe disease relevant human cells at scale. This is especially important for the neuroscience field as the central nervous system is not easily accessible for cellular biopsies and growing large amounts of patient neural cells is not possible. In addition to growing vast quantities of neurons, we now can generate patient-specific cells that harbor disease relevant genetic backgrounds, potentially leading to novel mechanism(s) responsible for the observed phenotypes.
Since the advent of this technology, numerous studies have been reported peering into a wide range of neurodegenerative disorders, such as Parkinson’s disease, Alzheimer’s disease, and Amyotrophic Lateral Sclerosis. Neurodevelopmental disorders have also been modeled and can be used as a bellwether for newly discovered disorders such as HNRNPH2-linked Bain Syndrome.
(1) To date there have been no peer reviewed reports of human stem cell disease modeling for Bain-type Syndrome. To our knowledge, generating patient-specific cell lines will be a first for the p.R114W variant. (2) Comparing these newly generated lines with the HNRNPH2 NLS variant patient-specific iPSC lines recently received from the Simons Foundation for Autism Research Initiative, will complement each other and enrich the entire research study. We are hopeful that rigorous investigation into the biological mechanisms of the p.R114W variant will yield new avenues for future therapeutic interventions. (3) Targeting rare neurodevelopmental monogenetic disorders with therapeutic oligonucleotide/genome editing approaches are still in its infancy with many opportunities. Successful treatments may act as a bellwether for more common neurological disorders such as autism and intellectual disability. (4) Since Drs. Bain and Chung discovered this rare disorder, they have developed and fostered a passionate community of both patients and caregivers which will continue into the foreseeable future. This puts the Columbia clinical and research teams in a unique position to lead in both diagnosis and research for this ultra-rare disorder. (5) Finally, there has been basic research carried out on the general biology of the RNA-binding protein family, their splicing regulation, tissue specificity and evolutionary conservation. We could therefore look to recruit new research collaborators interested in studying HNRNPH2 and its mutational consequences. We have reached out to Dr. Kinji Ohno from Nagoya University in Japan to gather his expertise on RNA-binding proteins. We plan to further expand our team of experts by collaborating with Dr. Stephen Floor at the University of California San Francisco who has worked with rare diseases and studies RNA-protein interactions and the effect of RNA on gene expression.
Consideration for X-inactivation: Since females have two copies of the X-chromosome while males only have one, dosage compensation is the process by which one of the two copies of the X-chromosome in females is randomly silenced. This achieves equal gene dosage for X-linked genes between the two genders. This also implies that additional care needs to be taken as it causes some problems regarding which X allele is expressed in reprogrammed iPSCs. It is of sufficient concern that the generated iPSCs carry the necessary background and, thus, they must be screened properly for X-linked inactivation.
Consideration for Quality Control: Upon recommendation from Dr. Chris Ricupero, we also propose using a well-established biobank facility (such as IBX) to store and distribute the iPSCs. Additionally, we need to have a third party facility (such as IBX) check and validate the iPSCs created by our cell programing originator, Applied Stem Cell, for pluripotency, karyotyping, and X-chromosome inactivation.
Differentiation into Neural Stem Cells & Synaptically Active Neurons: Dr. Davor Lessel recommended doing all transcriptomic and phenotyping studies in differentiated neurons to screen most accurately for neuronal phenotypes in the most relevant gene expression profile. Thus, to investigate the predicted deleterious effects the missense variants produce within the nervous system, we recommend differentiating the p.R114W isogenic iPSCs and the control iPSC line into neural lineages. The type of neural defects underlying X-linked Bain-type Syndrome is still an open question as to exactly when developmental problems arise and the specific cell type(s) most affected. Further, we do not know if the R114W mutation affects inhibitory, excitatory neurons or other neuronal cell-types. Therefore, it is necessary to investigate both early and later stage temporal time periods and various neuronal cell types. To this effect, our current recommendation is to work with Boston Children’s Neuron Core Facility to generate patient-derived neuronal cells that will contain a mixed population of differentiated cortical neurons. This process is estimated to take about 2 months and will produce a good yield of about 60 million neurons.
Basic Science 2: Identify the function of HNRNPH2 and its downstream targets by using NGS technologies (genomics, transcriptomics, and proteomics) in an effort to define HNRNPH2 variants.
Since HNRNPH2 is a member of a large RNA binding protein family, it is anticipated to be involved in a wide range of molecular and biochemical activities and its predicted pathogenicity may be wide ranging. In order to understand the mutational background of HNRNPH2 as well as identify the downstream targets of HNRNPH2, we suggest a two-pronged approach of using whole genome sequencing (WGS) and transcriptomics (RNA-seq).
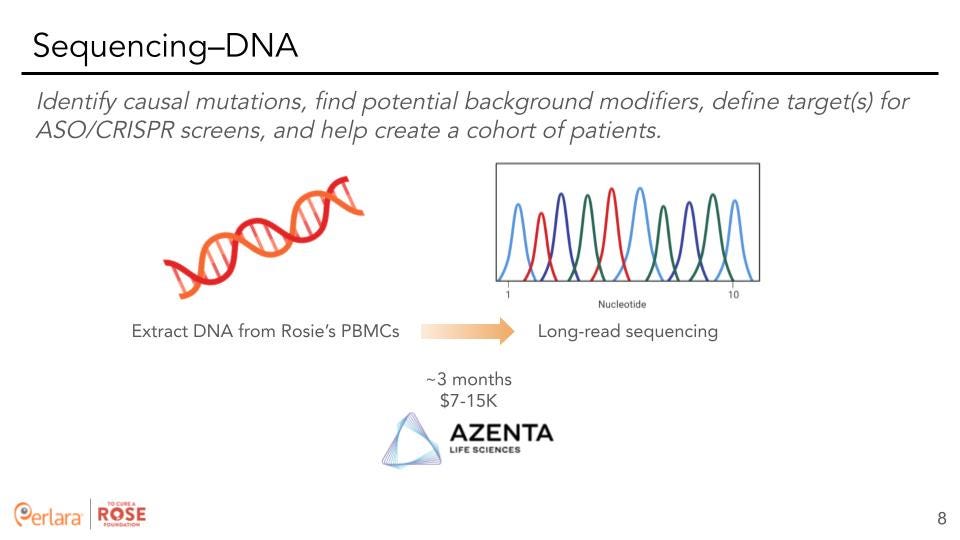
Whole genome sequencing: We already have a reasonable understanding of the mutational burden on HNRNPH2 based on sequence data (exome or targeted) from tens of affected individuals. Since HNRNPH2 is X-linked and affects males and females, it would be beneficial to learn if there are modifying mutations elsewhere in the genome that potentially modulate the effect of HNRNPH2. Specifically for TCAR, carrying out WGS would provide the ability to design ASOs in a more precise manner as we can consider Rosie’s haplotype when estimating ASO efficacy. Since Rosie has received an exome sequencing via GeneDx, our recommendation would be to communicate with GeneDx to verify if the exome sequencing was reflexed from a WGS backbone. Further, we recommend sending Rosie’s PBMCs from Applied Stem Cell to Azenta for WGS.
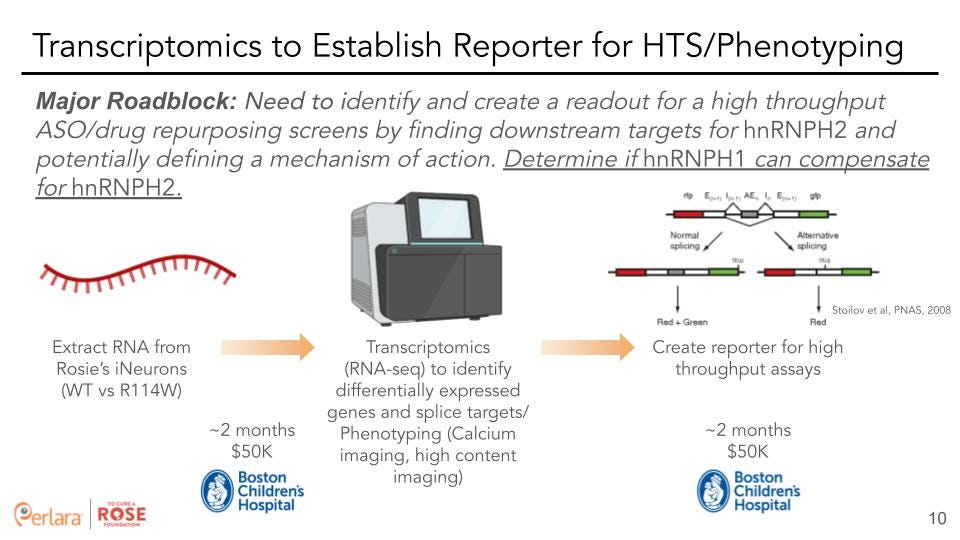
Transcriptomics to identify downstream targets: To identify the downstream targets of HNRNPH2 it will be beneficial to perform genome wide transcriptomics. Source material is a critical component for any transcriptomics analysis and differences in expression of genes and their isoforms across human tissues is a major consideration when evaluating a gene for targeted therapy. Importantly, each cell type is known to have a unique gene expression profile that includes expression of tissue specific isoforms and alternative splicing. Hence, a major barrier for RNA-seq is the requirement for a disease-relevant source material. While biopsies from affected tissues are the most ideal material, they are only available in limited settings. As an example, RNA sampled from blood does not adequately represent the transcriptomes necessary for the analysis of many rare disorders. Diagnostic biopsies are likely the best tissue source for RNA-seq, but are not available in many cases. Therefore a key consideration when carrying out RNA-seq analysis is the availability of “disease-relevant” tissue and its transcriptome.
In the case of HNRNPH2, the most relevant tissue type is the human brain that is matched to the ideal developmental stage. As this is not feasible, our recommendation (based on conversations with Dr. Lessel and others) is to carry out RNA-seq on neuronally differentiated cells. As it is currently unclear which neuronal cell type would be ideal, we further recommend that the initial sequencing be carried out in a mixed population as well as with control populations. It would be prudent to sequence (at least in triplicate to minimize technical and biological variation and provide statistical power) each of the cell-types (controls or samples, homozygous and hemizygous, iPSC and neuronal) so that pairwise comparisons of expression difference and splicing changes can be characterized. While approximately 20 Million reads (150nt paired-end) per replicate/sample would be sufficient for evaluating gene expression differences, in order to get a good estimate of changes in splicing, it might be necessary to sequence to a greater depth (at least 50 million reads). After generating the differentiated neurons, we recommend partnering with Boston Children’s Neuron Core Facility to perform transcriptomics as well. Although they do offer a fee-for-service platform, TCAR could also appeal to the “steering committee” at Boston Children’s to be accepted as a more intellectually involved project where a team of researchers would help TCAR phenotype the differentiated neurons and develop a screenable assay. Boston Children’s prefers to focus on projects that will help more than one patient, to which we could share data on the number of patients affected by a mutation in HNRNPH1/2.
Proteomics to identify interactors: Dr. Lessel’s team showed via functional characterization that the three most common HNRNPH2 missense variants revealed dysfunctional nucleocytoplasmic shuttling of proteins harboring the p.(Arg206Gln) and p.(Pro209Leu) variants, located within the nuclear localization signal, whereas proteins with p.(Arg114Trp) showed reduced interaction with members of the large assembly of splicing regulators (LASR) [7]. It would be beneficial to identify these interactions (in wild-type cells) and importantly, the consequent changes to these interactions in different mutational backgrounds. Talus Bio (
https://www.talus.bio/
) has developed a system combining nuclear sub-fractionation (i.e. separating nuclear proteins and complexes into groups) followed by quantification of the proteins in each group using mass spectrometry. This will provide answers to three related questions: a) what are the the proteins interacting with HNRNPH2; b) how these interactions are modified or disrupted when HNRNPH2 is mutated; and c) provide additional possibilities to design reporter assays involving HNRNPH2 and its interactors to build on the GFP work carried out in Dr. Lessel’s publication [7].
Data analysis: Standard downstream analysis of the data will provide a list of genes and transcripts that are either direct or indirect targets of HNRNPH2. These can be further classified into categories based on a set of rules: for example, a gene that shows consistent difference in expression and has a suspected HNRNPH2 binding site could be prioritized. Additionally, genes and transcripts could be grouped based on the pathways or functions they belong to. These data could be used to determine if HNRNPH1 plays a compensatory role in HNRNPH2 pathophysiology. Finally, based on the outcome, one or a few candidate genes could be identified for creating reporters for cellular screening of ASO and drug repurposing assays.
Basic Science 3: Develop an in vivo model for HNRNPH2-related NDD by collaborating with Jackson Lab to create mouse models carrying humanized versions of HNRNPH2.
Vertebrates, such as mice, offer a very attractive alternative as a disease model to study human development. Mouse models represent convenient tools to understand how specific mutations affect individuals as these introduced variants result in a mouse strain exhibiting some or all of the clinical phenotypes. By using the most appropriate mouse model, designing the treatment regimen that mirrors the clinical plan, and combining in-life repeated measurement of clinically-relevant phenotypes to post-mortem pathology analysis, preclinical researchers can ascertain critical insights into the efficacy and safety of the candidate treatment, refine it (if necessary), and increase its possibility of success in the clinical setting.

TCAR is currently working in collaboration with Jackson Laboratory to create four different mouse strains.
Insertion/replacement of WT human sequence into the mouse Hnrnph2 locus
Insertion/replacement of human R114W variant sequence into the mouse Hnrnph2 locus
Insertion/replacement of human R206W variant into the mouse Hnrnph2 locus
Deletion of the mouse Hnrnph2 sequence (null allele)
From its preliminary engineering attempt, Jackson Laboratory has identified 1 potential founder (carrier) for the R114W variant and 5 potential founders (carriers) for the R206W variant, and no carriers of the WT variant. They are currently in the process of repeating the attempt to generate the WT allele since there were no founders ID’d as well as the R114W allele since there was only 1 founder identified. Both injection attempts are in their model generation pipeline for reinjection and they should know by the end of April on the success of those repeat attempts.
With potential founders identified mid-January, the next step which is underway, is to set those animals to breed and then expand/establish a colony to continue to work with. As each generation takes at least 3 months, they are about 3-6 months out from establishing and expanding the colony for much validation work.
Once colonies are established (gene targeting confirmed via sequencing at the locus of interest) (and there can be lots of variables that can push out timelines relating to breeding fitness, etc.) they will proceed with phenotyping the mice. The first tasks would be around evaluating protein expression for each allele: establishing the baseline and determining functional outcomes of the new allele(s) as compared to the inbred strain or other relevant control. If Western blotting is unsuccessful, gene expression (mRNA or pre-mRNA) can be assessed. For each model there is typically a defined set of phenotyping assays to establish the model's relevance for human disease. In some cases there might be a biomarker that can be used to assess consequences of the patient mutation. To get the model ready for any sort of therapeutic testing, a baseline must be established for the desired assay along with a measure of improvement in response to therapy. Sometimes the assays are obvious, sometimes not.
It is worth noting that Jackson Laboratory previously created a HNRNPH2 knockout mouse which exhibited strange hair patterns and abnormal vocalizations, which is remarkably resonant with the loss of speech observed in patients. Jackson Laboratory intends to further perform a battery of behavioral tests along with transcriptomics to compare the WT with the HNRNPH2 knockout mice.
Invertebrate
Zebrafish have one ortholog (called hnrnph1) and there are two available zebrafish strains sa45741 and la012641Tg. The former is a point mutation that introduces a premature stop and the latter is an insertion. According to zfin (www.zfin.org) neither have discernible phenotypes and would therefore not be useful candidates for screening.
Interestingly, roundworms (C. elegans) have an ortholog for hnrnph1 (and not h2). There is one strain available from the Caenorhabditis Genomics Consortium (CGC) with a reporter, but it contains other genes (myo-2, unc-54) in the mutational background. We therefore feel that a worm model may be too complicated and not quite relevant to use for drug repurposing.
Basic Science 4: Reporter assays – Synthesizing the results from BS1 and BS2 to build dual color reporters that can be utilized for drug and ASO screens. Additionally, search for other high-throughput screenable phenotypes.
Alternative splicing has emerged as a promising therapeutic target in a number of human disorders. However, the discovery of compounds that target the splicing reaction has been hindered by the lack of suitable high-throughput screening assays.
Conversely, the effects of known drugs on the splicing reaction are mostly unclear and not routinely assessed. Finding compounds that modulate alternative splicing requires effective high-throughput screening assays. Small molecules that alter the splicing of particular exons have been identified by using several strategies, including RT-PCR, reporters producing luciferase or GFP, and a topoisomerase I phosphorylation assay. Each of these assays has limitations in the high-throughput screening of large chemical libraries. RT-PCR is costly and scales up poorly. Most in vivo splicing reporters have poor dynamic range or do not distinguish compounds affecting splicing from those altering transcription or translation.
Two recent studies demonstrate the utility of dual color reporter systems in improving the dynamic range and discriminating changes in alternative splicing from changes in transcription or translation [8,9]. These systems may require modification of a test exon to adapt it to the reporter, which may change its regulatory properties.
Stoilov et al [10] have developed a sensitive dual-reporter system for alternative splicing that can accommodate most cassette exons and many alternative 5′ and 3′ splice sites. They report the implementation of a design, which uses red and green fluorescent proteins (RFP and GFP) as reporters to screen chemical libraries for molecules that modulate the splicing of MAPT exon 10.
To screen for compounds that alter MAPT exon 10 splicing, they created a stable HEK293 cell line expressing the MAPT exon 10 reporter. This cell line showed partial inclusion of exon 10 and the expected fluorescent signals. They used this line to screen two libraries: a 1,100-compound library (Prestwick) of Food and Drug Administration (FDA)-approved and other drugs and a smaller 340-compound library (BioMol) consisting of enzyme inhibitors and ion-channel antagonists. After plating in 384-well plates, the cells were incubated with the BioMol library for 20 h or with the Prestwick library for 48 h and the fluorescence intensities were determined.
Similarly, Diez et al [11] have established a cell-based platform using metastatic melanoma cell line WM266-4 expressing hnRNPH2 conjugated with green fluorescent protein to enable assay development and screening. A High Content Screening assay was developed and validated in 384-well plate format, followed by miniaturization to 1,536-well plate format. This assay is a robust HTS-amenable high content screening assay capable of monitoring down-regulation of hnRNPH2. This assay is thus capable of identifying authentic down regulators of HNRNPH1 and HNRNPH2 in a large compound collection and, therefore, is amenable to a large-scale screening effort.

We recommended using either of these established assays or to modify these specifically with the downstream targets identified from the transcriptomics carried out with personalized cell lines.
We also recommend performing other phenotyping assays on the patient-derived differentiated neurons such as multi-electrode array, calcium imaging, and high content screening with live cell imaging to determine if there are other high throughput screenable phenotypes. Boston Children’s Neuron Core Facility has the ability to perform these screening assays and to aid in assay development on a high throughput scale. Ideally, the cell screening assays could be done in a more economical cell line than patient-derived neurons, such as engineered generic cell lines or patient-derived fibroblasts. Thus, it is worth performing a few phenotyping tests in these cell lines, such as transcriptomics or staining for cellular structures associated with RNA processing, to see if these cell lines would be valuable for initial drug and ASO screening. Rosie’s fibroblasts are already banked at the Manton Center.

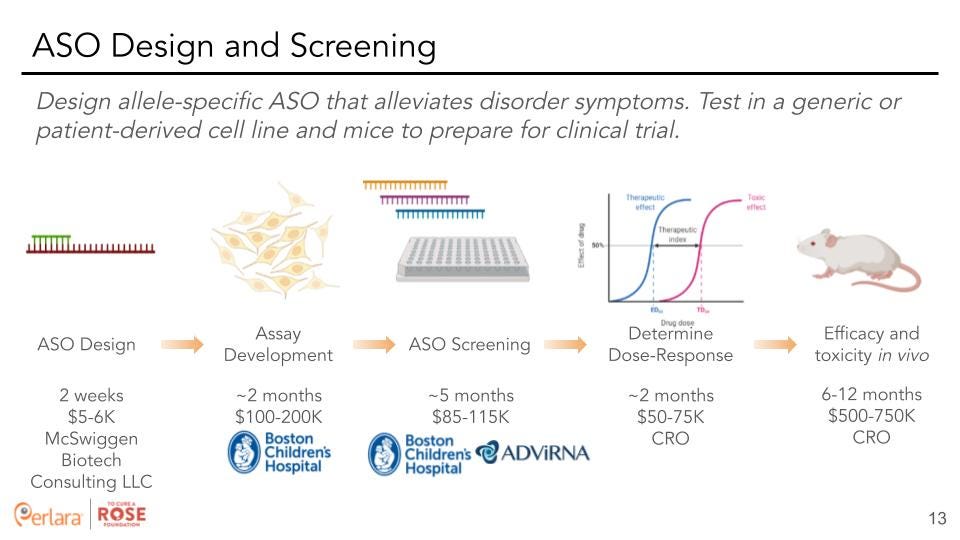
Therapeutic Track 1: ASO design – Design allele-specific and allele-agnostic ASOs to target HNRNPH2 variants and test these in in vitro and in vivo models.
Therapeutic Objective: Based on the proposed change of function of the HNRNPH2 variants, we propose to precisely target and knockdown the p.R114W allele-specific HNRNPH2 mRNA to slow down or reverse the pathogenic phenotypes in p.R114W iPSCs, neural precursor cells, and neurons. There have been two FDA approved medicines Spinraza (nusinersen), Tegsedi (inotersen) and over 40 ASOs in development [12-15].
ASO Functional Pathways: ASOs are small single-stranded nucleic acid polymers that bind RNA through Watson-Crick base pairing. ASOs fall into two functional categories with respect to gene down-regulation (versus gene up-regulation): initiating the endogenous RNAase H1 pathway for mRNA transcript cleavage and degradation, and steric hindrance that blocks ribosome binding, disrupting translation initiation, or modulating splicing that can restore protein expression through exon skipping [14,15]. For our specific needs, we will design ASOs to recruit and initiate the RNAse H1 machinery for HNRNPH2 mRNA cleavage and degradation, specific to the p.R114W missense variant. We hypothesize that substantial degradation of the proposed pathogenic HNRNPH2 protein will rescue the pathogenic phenotype predicted in the p.R114W iPSC derived neural cells.
ASO design and production: Although ASO binding concepts are straightforward, proper cellular uptake, mRNA secondary structure, binding affinity, and stability are all critical. Of interest, HNRNPH1 is a paralog of HNRNPH2 with only 15 amino acids separating them. However, their mRNA sequences are sufficiently divergent so we expect to design ASOs specific for HNRNPH2 while avoiding HNRNPH1 off-targets. Modifications to enhance stability and avoid degradation will be added to the ASOs once the initial candidate screening has been completed (23-25).
We recommend partnering with Jim McSwiggen Biotech Consulting LLC to design the top ASO candidates. Once Rosie’s WGS is complete, we recommend sending her sequencing data from Azenta to Jim McSwiggen to allow him to design SNP-specific ASOs. Previously, we sent him the human HNRNPH2 sequences that were inserted into the mice at Jackson Laboratory with the intent to design ASOs that will be applicable in both the in vivo and in vitro models. Getting the current research update from Dr. Paul Taylor’s lab at St. Jude’s will help us decide whether to pursue an allele-specific vs allele-agnostic ASO. Additionally, Dr. Davor Lessel noted that patients with nonsense mutations in HNRNPH2 appear to have less severe phenotypes than those with missense mutations. This observation adds weight to the argument for designing an allele-agnostic ASO, which would also be more applicable to a wider patient population.
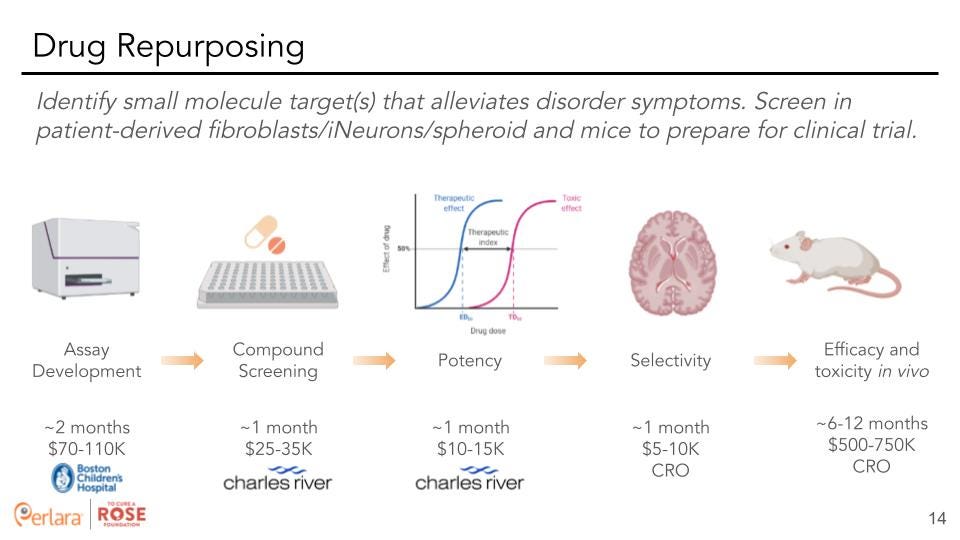
Therapeutic Track 2: Drug repurposing – Screen for small molecules that alleviate variant-specific phenotypes in cells.
Drug repurposing is defined as identifying a new use for an already approved drug, be it a century old drug or a drug that was just recently approved. This is an attractive option for multiple reasons. First of all, FDA-approved drugs won’t need to be de-risked for use in humans to the same degree as novel compounds. This will significantly cut development timelines for new treatments using an already FDA-approved drug to target a rare disease. Additionally, many of these drugs are produced commercially and are available at a fraction of the costs of a novel compound. We think that the short timelines and cost-efficient nature of drug repurposing screens should make this one of the key strategies for finding a possible therapy.
Identification of a pharmacological control for High-content screening (HCS) assay. Diez et al conducted a screen of a FDA-approved drug library (1,400 compounds) to identify a pharmacological control for HCS assay. Using their protocol, they found 8 hits (0.5% raw hit rate) of which they were able to purchase six compounds. Out of six commercially available compounds, four confirmed activity in dose response format (0.66% confirmed hit rate), namely dronedarone, mitomycin C, lapatinib free base form, and sunitinib. Two compounds exhibited EC50 values <20μM (0.07% high activity hit rate) (dronedarone = 17μM and mitomycin = 7.3μM). Examination of images of WM266-4-H2-GFP cells in presence of mitomycin showed a dose-dependent decrease of fluorescence and nuclei shapes suggesting that HCS assay is capable of correctly recognizing and quantitating the objects in the wells. Interestingly, none of the FDA approved melanoma drugs (trametinib, dabrafenib, vemurafenib, dacarbazine) decreased hnRNPH2 signal further corroborating their hypothesis that hnRNPH2 controls pathways previously unexplored for melanoma therapy.
We suggest a similar approach with either the cell-line established by Diez et al, or by using a dual color reporter (as established from BS4) transfected into a cell-line followed by screening drug libraries of interest to determine potential drugs of interest. We recommend hiring Charles River Laboratories for their extensive experience in small molecule screening. Additionally, Rarebase would be another potential CRO for drug screening. While screening in differentiated neurons (or a spheroid) would be most ideal, this model will also be much more costly than screening in patient fibroblasts. Thus, the decision of which cell type to utilize should be made after further evaluation of transcriptomics and screening for various phenotypes in the differing cell types.
References
Coe BP, Stessman HAF, Sulovari A, Geisheker MR, Bakken TE, Lake AM, et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet. 2019;51(1):106–16.
Khalil B, Morderer D, Price PL, Liu F, Rossoll W. mRNP assembly, axonal transport, and local translation in neurodegenerative diseases. Brain Res. 2018;1693(Pt A):75–91.
Purice MD, Taylor JP. Linking hnRNP function to ALS and FTD pathology. Front Neurosci. 2018;12
Fu X-D, Ares M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15(10):689–701.
Bain JM, Thornburg O, Pan C, Rome-Martin D, Boyle L, Fan X, Devinsky O, Frye R, Hamp S, Keator CG, LaMarca NM, Maddocks ABR, Madruga-Garrido M, Niederhoffer KY, Novara F, Peron A, Poole-Di Salvo E, Salazar R, Skinner SA, Soares G, Goldman S, Chung WK. Detailed Clinical and Psychological Phenotype of the X-linked HNRNPH2-Related Neurodevelopmental Disorder. Neurol Genet 2021;7:e551. doi:10.1212/NXG.0000000000000551
Bain JM, Cho MT, Telegrafi A, Wilson A, Brooks S, Botti C, Gowans G, Autullo LA, Krishnamurthy V, Willing MC, Toler TL, Ben-Zev B, Elpeleg O, Shen Y, Retterer K, Monaghan KG, Chung WK. Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. AmJ Hum Genet. 2016 Sep 1;99(3):728-734. doi: 10.1016/j.ajhg.2016.06.028.
Kreienkamp HJ, Wagner M, Weigand H, McConkie-Rossell A, McDonald M, Keren B, Mignot C, Gauthier J, Soucy JF, Michaud JL, Dumas M, Smith R, Löbel U, Hempel M, Kubisch C, Denecke J, Campeau PM, Bain JM, Lessel D. Variant-specific effects define the phenotypic spectrum of HNRNPH2-associated neurodevelopmental disorders in males. Hum Genet. 2022 Feb;141(2):257-272.
EA Newman, et al., Identification of RNA-binding proteins that regulate FGFR2 splicing through the use of sensitive and specific dual color fluorescence minigene assays. RNA 12, 1129–1141 (2006).
JP Orengo, D Bundman, TA Cooper, A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res 34, e148 (2006).
Stoilov P, Lin CH, Damoiseaux R, Nikolic J, Black DL. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc Natl Acad Sci U S A. 2008 Aug 12;105(32):11218-23. doi: 10.1073/pnas.0801661105. Epub 2008 Aug 4.
Diez J, Rajendrarao S, Baajour SA, Sripadhan P, Spicer TP, Scampavia LD, Minond D. Development and Pilot Screen of Novel High Content Assay for Down Regulators of Expression of Heterogenous Nuclear Ribonuclear Protein H2. Cell Physiol Biochem. 2021 May 19;55(3):265-276
Crooke ST (2021) A call to arms against ultra-rare diseases. Nat Biotechnol 39:671-677.
Goyal N, Narayanaswami P (2018) Making sense of antisense oligonucleotides: A narrative review. Muscle Nerve 57:356-370.
Liang XH, Nichols JG, De Hoyos CL, Crooke ST (2020) Some ASOs that bind in the coding region of mRNAs and induce RNase H1 cleavage can cause increases in the pre-mRNAs that may blunt total activity. Nucleic Acids Res 48:9840-9858.
Roberts TC, Langer R, Wood MJA (2020) Advances in oligonucleotide drug delivery. Nat Rev Drug Discov 19:673-694.