KCNH1 Cure Roadmap
The 4th Cure Roadmap was commissioned by an entrepreneurial family-led foundation based in San Diego. Our core recommendations are drug repurposing and both allele-agnostic and allele-specific ASOs.


KCNH1-Associated Neurodevelopmental Disorders Cure Roadmap
Prepared for Cure KCNH1 Foundation by Perlara PBC
January 2022
VISION
The goal of this Roadmap is to identify ways in which children with pathogenic KCNH1 variants may maximize their growth potential, especially in the cognitive domain but also in the motor domain and with minimization of epilepsy symptomatology, and for them to go on to live happy, healthy, and complete lives. This multi-year multi-modality drug development plan investigates the academic and commercial scientific landscape in order to strategize rational therapeutic development, anticipating the necessity of a small clinical trial given the ultra-rare incidence. In addition, this document seeks to understand the potential market for tools and assets developed to target KCNH1 variants for the eventual transition to a biotech startup. The goals of developing a therapy for a single individual with a novel variant are weighed against the individual and societal benefits of production and preclinical testing of an allele-naive approach, in recognition that the biology will ultimately dictate the approach with the highest chance of success.
This Roadmap envisions the following milestones and timelines:
In 4-6 months, the preliminary experimentation will be performed to assess the likelihood for success of the proposed therapeutic modalities. Drug repurposing and antisense oligo (ASO)-based precision medicine modalities will be primarily tested, putting our theoretical assumptions about which treatment avenues offer the most long-term promise to the test. As the basic biology surrounding the role of KCNH1 in the brain is limited, especially the impact of haploinsufficient (50% reduction) or absent expression and function, we advocate for pursuing allele-specific and allele-agnostic ASOs in parallel until the impact of both can be directly compared in vitro.
In 7-12 months, we will have entered the validation stage of promising therapeutic modalities. In-depth characterization of ASOs and repurposed drug candidates will be underway. For KCNH1-associated neurodevelopmental disorders, this will primarily consist of the efficiency of knockdown in channel expression for an ASO vs the efficiency of channel block (i.e., reduced potassium conductance) for a repurposed drug. Given that KCNH1-associated neurodevelopmental disorder is still considered ultra-rare (i.e. reported in <30 individuals), recommendation to refer to n-Lorem has been made and we will plan to reach out to them to compare and combine design strategies where possible. If the ASO-based precision medicine modality fails as a therapeutic avenue in the first 7-9 months, we will pivot to developing a KCNH1-targeted protein degrader.
In 16-24 months, we will have multimodal, preclinical data for promising therapies in hand so that we can pursue fundraising and partnership investment opportunities with biotech and pharmaceutical companies, particularly in the neuroscience and oncology spaces. We will also have a pre-IND meeting with the FDA to vet drug development plans and evaluate the strength of existing natural history studies. It is at this stage that we will expand the pioneer “n-of-1” program to conduct pivotal clinical trials to serve the KCNH1 community writ large.
In 2-3 years, precise, targeted treatments informed by careful molecular in vitro and preclinical ex vivo analyses will be accessible to children and adults of all ages with KCNH1-associated neurodevelopmental syndromes. Standardized natural history study data and regulatory agency-endorsed clinical trial protocols will be accessible to researchers, clinicians, companies and patients via federated data-sharing platforms. Drug repurposing and clinical biomarker discovery supported by preclinical disease models and patient biosamples will augment the standard of care.
Where possible, we will use gains made in our understanding of the fundamental biological underpinnings of KCNH1 gain-of-function to complement or even reciprocally benefit the oncology sector, in which robust and nearly-universal ectopic expression of KCNH1-encoded hERG (Kv10.1) channel isoforms demarcates and intensifies the pathogenicity of malignancies.
AUTHORS
Tracy S. Gertler, MD PhD. Attending Physician, Lurie Children’s Hospital of Chicago; Assistant Professor of Pediatrics (Neurology), Northwestern University Feinberg School of Medicine; Cure Guide, Perlara
Whitney Dolan, PhD. Cure Guide, Perlara
Ethan O. Perlstein, PhD. CEO of Perlara PBC & Maggie’s Pearl LLC

REVIEWERS
Luis Pardo, MD PhD. Professor, Max Planck Institute for Experimental Medicine

EXECUTIVE SUMMARY
The KCNH1-associated Neurodevelopmental Disease Cure Roadmap marries the best of two complementary models of patient-driven drug development. Firstly, a mutation-agnostic approach that aims to identify therapies that normalize channel overactivity; and, secondly, a mutation-targeted approach that aims to reduce toxic channel hyperactivity in the CNS. A blended strategy is necessary because of the inability to predict the success of any one therapeutic strategy for treating KCNH1-associated Neurodevelopmental Disease.

Tristan’s family and the KCNH1-associated Neurodevelopmental Disease community are poised for capital-efficient, modality-diversified drug development. Although still considered an ultra-rare disease, there are key biological insights that have already been made about KCNH1 and the hEAG1 channel it encodes, including sequence, structural, and biochemical similarities to hERG channels as well as its ectopic expression in neoplastic cells. A limited number of heterologous cell expression systems and animal (e.g., zebrafish and mouse) models exist and can be expanded/supplemented on a variant-by-variant basis, but will all require additional characterization and comparison. In addition, a clear definition of quantifiable and near-universal clinical endpoints will be needed to define a standardized clinical trial protocol that can be presented to regulatory agencies and pressure-tested in single-patient “n-of-1” pioneer studies.
This Roadmap initiative was kicked off by Tracy Gertler, Whitney Dolan, and Ethan Perlstein in November 2021. During the next three months, we invited reviewers from academia, medicine, and industry to participate and have their voices heard. This Roadmap will continue to be transparently and rigorously reviewed “post-publication” throughout early 2022 as we begin experimentation on potential therapeutic avenues.
The goal of the KCNH1-associated Neurodevelopmental Disease Cure Roadmap is to stimulate development of medicines that will treat (and potentially cure) disability characterized by intellectual impairment, motor delays, and intractable epilepsy. Perlara’s agile Cure Guide operating structure and a Slack-first, globally distributed team will enable research capacity building and preclinical proof-of-concept studies across multiple programs that will ultimately coalesce into a biotech startup ecosystem urgently developing new curative medicines.
The KCNH1 Cure Roadmap project will deliver:
Exploration of variant-agnostic and variant-specific therapeutic modalities as potential treatments of KCNH1-neurodevelopmental disorders. These modalities include:
Multiple ASO strategies targeting pathogenic KCNH1 overactivity
An unbiased drug repurposing screen using HEK293 cells expressing KCNH1 carrying Tristan’s variant compared to a population control.
Outlining of a standardized clinical trial protocol that will be validated by regulators in order to lower the barrier to investment into specific therapeutic tracks.
Testing Imipramine for seizure control in a small cohort
Anticipating deliverance of an ASO intrathecally for KCNH1 knockdown
Disease models, prognostic tests, clinical trial protocols and disease-modifying therapies developed for KCNH1-associated Neurodevelopmental Disease may have broader applicability and impact across other ‘gain-of-function’ potassium channelopathies, including those closely related (e.g. KCNN3-associated neurodevelopmental disease) in which a similar mechanism and similar clinical phenotype suggest shared underlying pathophysiology, and may ultimately impact healthspan extension. This broader applicability will make investments into KCNH1-associated Neurodevelopmental Disease compelling beyond the stakeholders who are directly affected by the disease, even prior to considering a foray into the oncology domain.

INTRODUCTION
KCNH1-associated neurodevelopmental disorders
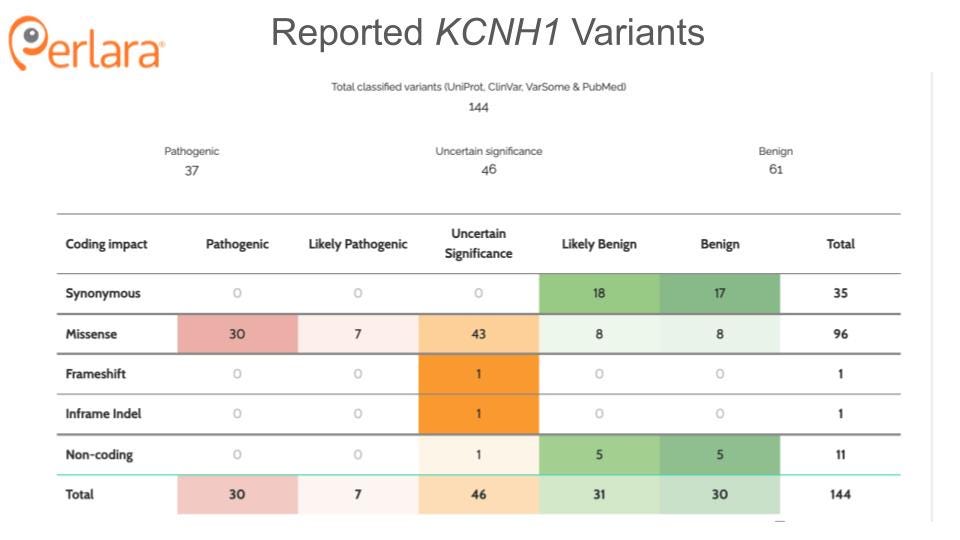
KCNH1, located on chromosome 1q32.2, encodes the voltage-gated potassium channel Kv10.1, also called an EAG1 (ether-a-go go) channel for its associated behavioral phenotype in Drosophila. Heterozygous, autosomal dominant germline variants, thus far predicted to be primarily gain-of-function, have been noted in two related phenotypic presentations: Temple-Baraitser syndrome (TBS; OMIM #611816) and Zimmermann-Laband syndrome-1 (ZLS-1; OMIM #135500) (Mastrangelo et al. 2016; Gripp et al. 2021; Kortüm et al. 2015; Fukai et al. 2016; Mégarbané et al. 2016; Simons et al. 2015; H. Wang, Zhang, and Ding 2021). ZLS was first described based on clinical observations of gingival hyperplasia, distinctive facial features (large ears, soft tissue enlargement of the nose, skeletal changes, reduced nail size) and intellectual disability; as it has subsequently been discovered to be associated with convergent presentation of variants from three genes (i.e. phenotypic convergence with KCNN3 and ATP6V1B2 (Gripp et al. 2021). We herein only describe the subset associated with KCNH1. TBS has thus far been exclusively linked to mutation of KCNH1. Clinical features of TBS include intellectual disability, anomalies of the first ray of the upper and lower limbs with absence/hypoplasia of the nails, seizures, and characteristic facial features. The significant overlap of clinical features between TBS and ZLS, and the observation that not all individuals with KCNH1-associated epilepsy exhibit the characteristic features of these syndromes suggests that KCNH1-associated disorders likely exist on a continuum (Aubert Mucca et al. 2021; Bramswig et al. 2015).

Only a handful of individuals with mutations in KCNH1 have been reported in the literature to date. The majority of these mutations are novel, de novo variants though increased reporting has identified several recurrent variants. Individuals with pathogenic variants in the pore region of the protein tend to display more severe phenotypes than those with mutations outside of the pore (von Wrede et al. 2021). Tristan is a two year-old boy who carries a heterozygous, de novo mutation in KCNH1 (c.1678C>T; p.P560S); this amino acid is located in the intracytoplasmic C-terminus, outside of any known functional domain. Notably, proline is evolutionarily conserved at this position in human, mouse, rat, frog, fish, fly and worm, as shown here:

He is developmentally delayed and has mild epilepsy characterized by febrile seizures. At 15 months old Tristan was diagnosed with Zimmermann-Laband Syndrome. Recently, Tristan’s seizures have been well-controlled with anti-eplieptic medication. Consistent with his more mild phenotype, Tristan’s mutation is in the C-linker region of KCNH1. Heterologous expression experiments have confirmed that Tristan’s mutation acts as a gain-of-function (L. Pardo, personal communication).
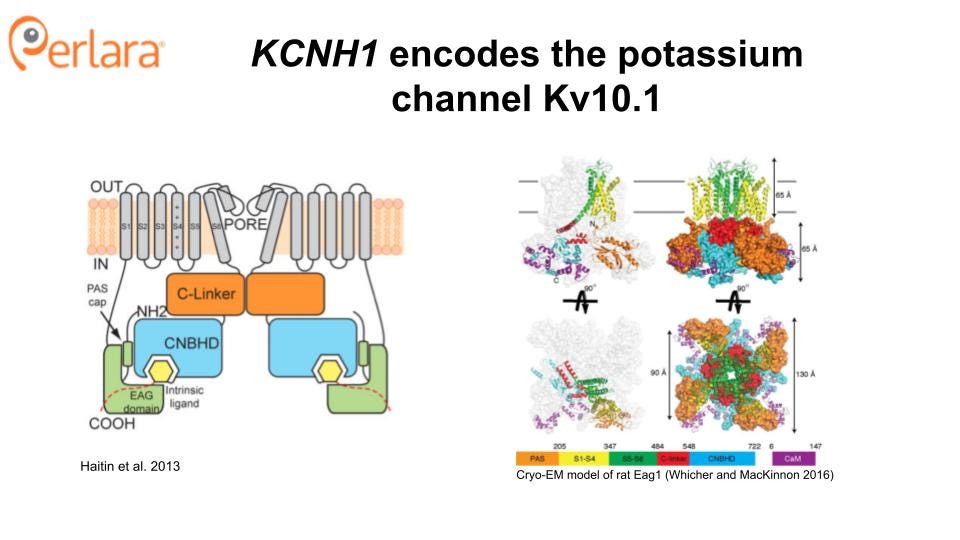
KCNH1 Structure and Function
KCNH1 is one of the seven members of the KCNH gene family. KCNH1 encodes the ion channel hEAG1 (also known as Kv10.1), a homotetrameric complex, with each subunit comprising an N-terminal Per-Arnt-Sim (PAS) domain, six transmembrane domains, a C-linker region, and a cyclic nucleotide-binding homology (CNHB) domain (Haitin, Carlson, and Zagotta 2013; Whicher and MacKinnon 2016). Transmembrane helices S1-S4 form the voltage sensor domain, and helices S5 and S6 form the pore of the channel. Notably, Kv10.1 has strong structural similarity to the cardiac potassium channel hERG1, also known as Kv11.1. This is important because inhibition of hERG1 can lead to a potentially fatal cardiac arrhythmia (Torsade de Pointes). Thus, any small molecule therapeutics developed against hEAG1will also need to be counter-screened against hERG1. Overcoming hERG1 liability is likely to be a significant challenge, as no small molecule inhibitors selective for hEAG1have been identified to date though multiple labs have reportedly attempted to do so (Toplak et al. 2022, 2021). However, as a large number of hERG blockers have already been identified, these small molecule libraries may serve as starting points for structural modification in parallel with an unbiased screen. The identification of a specific spider toxin for hEAG1 (Ma et al. 2018) is also an encouraging addition to the toolset needed to identify a small molecule channel blocker.
In addition to regulation by assembly, hEAG1 is also regulated by cellular ion concentrations, second messengers, and interactions with other proteins (Urrego et al. 2016; Han et al. 2016, 2017; Bronk et al. 2018; Hsu et al. 2017; Fang et al. 2021). The channel undergoes a high rate of turnover and is found at both the plasma membrane and the inner nuclear membrane (Kohl et al. 2011; Y. Chen et al. 2011). Calmodulin, in its calcium-bound state, interacts with hEAG1 to promote channel closure (Schönherr, Löber, and Heinemann 2000). Glycosylation of two asparagine residues is required for trafficking to the plasma membrane (Napp et al. 2005). Together, those facts suggest that it will be possible to identify modifiers (suppressors and enhancers) of hEAG1 channel function that target upstream or downstream proteins.

Physiological Role of KCNH1
The physiological role of hEAG1 is not well understood. The channel is primarily expressed in the brain, where it appears to limit the excitability of neurons when stimulated to fire at high frequencies (Ufartes et al. 2013). This is counterintuitive to the observation that gain-of-function in a potassium channel leads to epilepsy (Niday and Tzingounis 2018). Although one can propose a number of hypotheses to explain this apparent discordance, understanding the mechanisms of pathogenesis of the gain-of-function variants thus far identified in KCNH1 is limited by the fact that they have only been studied in heterologous expression systems, and have not been characterized in any whole-animal models. For example, development and characterization of a mouse (and even fly and worm avatars) carrying the equivalent of Tristan’s variant will enable greater understanding of the pathophysiology of KCNH1 gain-of-function variants and will be an essential resource for preclinical testing of potential therapeutics. Zebrafish and/or patient iPSC-derived neurons represent potential other neuronal preclinical models.
In addition to its role in neurons, hEAG1 also contributes to cell cycle progression and proliferation, both in normal cells (J. Zhang et al. 2012; Y.-Y. Zhang et al. 2014; Urrego et al. 2016) and in cancerous cells. In normal cells, KCNH1 is only expressed during the G2/M transition (Urrego et al. 2016), when it contributes to the disassembly of primary cilia (Sánchez, Urrego, and Pardo 2016). Several observations suggest that some of the developmental features of TBS and ZLS may be related to the effects of hEAG1 on cilia formation and resorption. First, some of the clinical features of TBS and ZLS are similar to those commonly observed in ciliopathies (oral, facial, and digital abnormalities, intellectual disability). Second, transfection of mouse embryonic fibroblasts with wild-type KCNH1 causes a reduction in the percentage of ciliated cells, and transfection with KCNH1 carrying a gain-of-function mutation known to cause ZLS exacerbates this effect. Inhibition of hEAG1 in hTERT-RPE1 cells with the antifungal agent (and nonselective channel blocker) astemizole had the opposite effect, suggesting that the role of hEAG1 in cilia formation is linked to the canonical, potassium-conducting activity of the channel (Sánchez, Urrego, and Pardo 2016).
KCNH1 is overexpressed in 70% of cancers (Hemmerlein et al. 2006). Expression of KCNH1 has been associated with tumor cell growth and invasion, as well as poor prognosis (Ouadid-Ahidouch, Ahidouch, and Pardo 2016; Cázares-Ordoñez and Pardo 2017; X. Wang et al. 2017; J. Chen et al. 2021; Li et al. 2017; Chávez-López et al. 2020; L. A. Pardo et al. 1999). Importantly, inhibition of hEAG1 has been shown to slow the proliferation of cancer cell lines, making it an appealing therapeutic target for the treatment of certain types of cancer (Wu et al. 2012; Hui et al. 2015; Weber et al. 2006; Luis A. Pardo and Stühmer 2008; Gómez-Varela et al. 2007; X. Wang et al. 2019; Bai et al. 2013; Wulff, Castle, and Pardo 2009; Z.-J. Wang et al. 2020). As such, the therapeutics proposed in this Roadmap have a potential dual use in the oncology sector, which is likely to garner additional interest from biotech companies.
A diversified portfolio of therapeutic modalities
The inability to predict with certainty the success of any one treatment modality necessitates a diversified portfolio of therapeutic approaches that are embodied in distinct but combinable therapeutic modalities - for example, an oral small molecule drug that blocks channel activity, and an intrathecal-delivered ASO to down-regulate channel expression. At some point to be determined in the future, a KCNH1 targeted protein degrader approach may replace the ASO strategy.
When considering the most clinically actionable therapeutic modalities for the treatment of KCNH1-related neurodevelopmental disorders, we considered three major questions:
How challenging is it to develop? Has it been tried before (successfully or unsuccessfully)? Are the necessary resources available, and if not, how long would it take to develop them? What are the regulatory requirements? Modalities that are particularly challenging to develop, or have special regulatory requirements will ultimately slow therapeutic development and delay treatment.
What are the predicted efficacy and safety of the modality? How likely is it to work and what is the likelihood of undesirable side-effects?
What is the size of the patient population? Is there a large enough cohort with a measurable endpoint to enable conclusions about the efficacy of a given therapeutic?
With these considerations in mind, the KCNH1 Cure Roadmap proposes two therapeutic tracks: (1) development of a mutation-agnostic ASO, and (2) drug repurposing. Both therapeutic tracks directly target the pathogenic overactivity of hEAG1, either by knocking down KCNH1 gene expression, or by molecular inhibition of the channel. Both tracks will also benefit from the development of a common set of resources, and will be informed by several preliminary experiments. Each track also presents its own unique advantages and challenges, such that parallel execution is recommended to quickly identify the most viable path forward.

The KCNH1 Community
Paramount to the success of the Roadmap will be to assemble a small cohort of patients with KCNH1-associated disorders. The KCNH1 Foundation, founded by Michaelle Jinnette and Kevin Witt, is expected to receive its 501(c)(3) status in the first quarter of 2022. The Foundation will serve as a nucleus for community and cohort building, as well as fundraising for the therapeutic tracks outlined by the Roadmap. The KCNH1 Foundation board includes Dr. Luis Pardo and Dr. Al George. Dr. Pardo is one of the world’s leading experts on KCNH1. He has studied the function of KCNH1, particularly in relation to oncology therapeutics, for over several decades. Dr. George’s work focuses on using electrophysiology to under rare channelopathies. He led the study that proved the causative link between KCNH1 and Temple-Baraitser Syndrome. Together, with the support of Perlara, the community is well-positioned to make rapid and significant impacts in our understanding of KCNH1-associated neurodevelopmental disorders and the development of therapeutics for their treatment.
THERAPEUTICS READINESS
The development of new medicines requires disease models, patient-derived biosamples and cells, and a deep mechanistic understanding of pathophysiology that identifies disease modifying drug targets, including the monogenic driver gene itself.
The pathophysiology of KCNH1-related neurodevelopmental disorders is not well understood; however, all of the disease-causing KCNH1 variants characterized to date are gain-of-function mutations (at least in recombinant cell lines), suggesting that the phenotypes likely ultimately arise from hyperactivity of hEAG1. Thus, any therapeutic approach aimed at ameliorating the effects of these mutations is best targeted directly to the potassium-conducting activity of the channel. Channel activity, and the inhibition thereof, can be measured in a variety of in vitro and in vivo model systems. Accordingly, cellular and animal models (cell lines, patient cells, animal models) can be employed to determine the degree to which a given intervention inhibits the function of the channel. Some of these resources are currently available, while others will need to be developed.
Evidence from knockout mice suggests that knockdown or inhibition of Kcnh1 will be well tolerated in humans. Mice lacking the voltage sensor and pore domains of Kcnh1 develop normally and exhibit normal memory, learning, socialization, and motor control, with only mild ADHD noted (Ufartes et al. 2013). Complete loss-of-function has not been observed in humans, and is presumed to be embryonic lethal; however, there is also no evidence for haploinsufficiency as a disease-causing state. Only one nonsense mutation has been reported in the literature. Of the three related individuals heterozygous for this variant, two are apparently healthy, while the third exhibits generalized epilepsy and intellectual disability (von Wrede et al. 2021). These observations suggest that a reduction in KCNH1 expression is tolerated in humans, supporting KCNH1 knockdown as a viable therapeutic direction. Developing animal models, both loss-of-function and gain-of-function, will be important for validating these assumptions.
Disease models
Cell-based and animal models of KCNH1 gain-of-function will enable greater understanding of the pathophysiology of KCNH1-related disorders, and will allow for the experiments necessary to identify potential therapies for those disorders. These systems each have their own distinct advantages and limitations, and will be employed strategically for efficient therapeutic development.
Below is a description of the state of currently available KCNH1 disease models:
Cell-based models
Xenopus laevis oocytes and HEK293 cells
Xenopus oocytes and HEK293 cells are heterologous expression systems commonly used to characterize the function of mutant ion channel proteins. These cells are inexpensive to maintain, easy to culture, and are amenable to the high-throughput assay development needed for drug repurposing screens and post-screening hit validation studies. Heterologous protein expression in these systems is robust, and channel function can be measured using fluorescence-based assays or electrophysiology readout. Tristan’s variant has already been expressed and characterized in Xenopus oocytes, where the channel was found to open at more hyperpolarized potentials and to deactivate at a slower rate; this is considered a gain-of-function, as these biophysical shifts result in an increased total potassium conductance.
Primary neuronal cell cultures
Primary neuronal cultures from rodent hippocampal and cortical explants can serve as a heterologous expression system wherein the broader effects of a particular channel variant on neuron function, development, and neuronal networks can be assessed. If the channel is natively expressed in neurons, a classic approach is to overexpress the channel using either electroporation or AAV-mediated delivery. One disadvantage of this approach is that expression levels can vary by cell and are difficult to control preemptively. The effects of KCNH1 gain-of-function mutants have not been examined in neuronal cell cultures but may be a reasonable next step to gauge the impact of an enhanced hEAG1-mediated conductance. Such experiments would be useful for understanding whether different gain-of-function variants have the expected impacts and whether different variants behave similarly. These experiments are not crucial for the success of the Roadmap, but might become more important if gain-of-function mouse models do not display the expected phenotypes. These cell cultures can also be adapted to higher-throughput screening methods, and might be particularly useful in the case that patient-derived cell lines are not available.
Patient-derived iPSCs
iPSC (induced pluripotent stem cell) lines along with isogenic controls can be generated from peripheral blood mononuclear cells (PBMCs), which are collected via a blood draw, or from primary fibroblasts grown in primary culture after skin biopsy. This process generally requires at least 2-3 months, and varies in cost from $2,000-15,000 based on academic vs commercial cores, extent of validation performed, and number of clones generated. Additional NIH-validated stem cell lines are available from the Coriell human tissue database if sex-matched controls are needed, though these are primarily from fibroblast and cord blood lineages.
The primary advantage for this technique is that the disease-permissive genomic variant and modifiers first identified in a child with a neurodevelopmental disease are preserved, and can be CRISPR-corrected to demarcate the contribution of a single pathogenic variant. However, differentiation into specific neuronal cell types, especially inhibitory as compared to excitatory neurons, is still a relatively challenging and slow procedure that is challenging to reliably reproduce. Patient-derived stem cell lines can be differentiated to produce a specific type of cell, or several types of cells, in order to study endogenous protein or gene function in a particular cellular context, or with regard to cellular activity. Patient-derived cell lines are particularly important for the development of therapeutic approaches that are intended to be allele-selective. iPSCs are currently in the process of being generated from Tristan’s cells.
Cancer cell lines
Given that KCNH1 is overexpressed in many cancers, cancer cell lines may offer a convenient resource for preliminary testing of therapeutic potential against KCNH1 gene expression or function. In later stages of post-Roadmap research activities, potential therapeutics can be screened in cancer cell lines for their anticancer activity (e.g. ability to inhibit the epithelial-to-mesenchymal transition) to determine whether they might have a dual use as cancer therapeutics.
KCNH1 is expressed in MCF-7 (breast cancer), HeLa (cervical cancer), SH-SY5Y (neuroblastoma), and EFM-19 (carcinoma), MDA-MB435S (breast carcinoma), HT1080 (fibrosarcoma), and A204 (rhabdomyoscarcoma) cell lines, and knockdown of KCNH1 using an antisense RNA reduced the proliferation of these cells (L. A. Pardo et al. 1999; Weber et al. 2006; Mello de Queiroz et al. 2006). KCNH1 is also expressed in IGR1 and IPC298 melanoma cell lines (Meyer et al. 1999), and incubation with imipramine reduced DNA synthesis, metabolism, and proliferation of IGR1 cells (Gavrilova-Ruch et al. 2002).
Cilia formation in RPE1, 3T3 and MEF cell lines
REP1, 3T3, and mouse embryonic fibroblast (MEF) cell lines are commonly used model systems for studying cilia formation, and KCNH1 mRNA is almost as abundant in hTERT-RPE1 cells as in brain cells (Sánchez, Urrego, and Pardo 2016). Given that gain-of-function in KCNH1 results in fewer ciliated cells, and that disruption of cilia formation may underlie some of the phenotypes of KCNH1-related disorders, therapeutic candidates could be tested for their ability to restore cilia formation in these cell lines as a proxy for whether we can expect a therapeutic to have actual therapeutic benefits. These cell lines are amenable to transfection, and MEFs can be generated from mutant mouse lines. Cilia formation can be monitored by image-based fluorescence screens. Interestingly, research in other neurodevelopmental diseases where microcephaly is a prominent phenotype have used iPSC-derived organoids to reproduce primary ciliation and centrosome assembly required for early neuronal proliferation (Gabriel et al. 2020). Once iPSC-derived cell lines are generated and validated, they can be maintained and expanded indefinitely for future experiments such as these.
Animal models
Mouse models
One of the main advantages of a transgenic KCNH1-knockin mouse is that variant-targeted medicines like nucleic acid therapies can be tested in an intact mammalian brain, as compared to a heterologous expression system, primary neuronal culture, or iPSC-derived neuron culture. If an ASO-based approach appears to be the most promising route, selecting target sequences that are orthologous in mice and humans (and rats as well) would have the benefit of using the same molecule for preclinical testing in rodents and initial clinical trials in humans.
Dr. Cas Simons’ lab has generated a mouse carrying the equivalent of the human recurrent p.I494V variant (also evolutionarily conserved from worms to humans), which has been observed in three individuals with TBS. The mice have not been extensively phenotyped, but heterozygous mice were apparently healthy, aside from anecdotally-observed decreased male fertility. It is possible though not yet tested that homozygous mice exhibit a more readily apparent phenotype consistent with KCNH1-associated neurodevelopmental disorders.
In addition to the mouse generated by the Simons lab, we propose development of a mouse carrying the equivalent of Tristan’s variant. However, given the location of Tritan’s variant in the C-linker region of the protein, there is a strong chance that the mouse will have an even more mild phenotype than Kcnh1 I494V mouse. If this is the case, we may need to consider generating a mouse with a different gain-of-function mutation that is predicted to have a stronger, quantifiable phenotype, such as seizure frequency, to test the efficacy of potential therapeutics.
We also propose development of a true Kcnh1-null mouse, to assess whether there might be adverse effects associated with the complete loss of expression, given the limited ability to titrate protein levels with current knockdown technologies (i.e. ASO, or targeted protein degrader). A knockout mouse previously described with a phenotype notable for only mild ADHD (Ufartes et al. 2013) was generated by deletion of the voltage sensor and pore domains alone, and therefore does not exclude the possibility of expression and non-canonical function of the remaining domains.
Zebrafish model
While knockout of Kcnh1 is well-tolerated in mice, complete kcnh1 loss of function in zebrafish (Danio rerio) results in growth retardation, delayed hindbrain formation, and embryonic lethality (Stengel et al. 2012). kcnh1 gain-of-function mutations have not been studied in zebrafish, but it may be possible that knock-in variants in either kcnh1a or kcnhb within the zebrafish genome could serve as a model system if they recapitulate differences in neurologic and/or craniofacial development. If gain-of-function mice do not display significant phenotypes, therapeutics could potentially be tested in a zebrafish model; the relatively shorter gestational period would also be advantageous for testing a greater number of compounds.
Fly and worm models
In parallel with zebrafish modeling activities, it may be worth paying a contract lab to develop a fly avatar and a worm avatar expressing Tristan’s P560S variant. These models could then be tested for expected gain-of-function behavioral phenotypes and compared to other gain-of-function mutants. Depending on the results and challenges associated with other disease modeling activities, fly and worm avatars may become useful for drug repurposing screens or for assessing the mechanism of action and tolerability of hits from cell-based drug repurposing screens that are described below.
THERAPEUTIC TRACKS
We propose two therapeutic tracks, which will be explored in parallel.
THERAPEUTIC TRACK 1: TARGETED KNOCKDOWN OF KCNH1
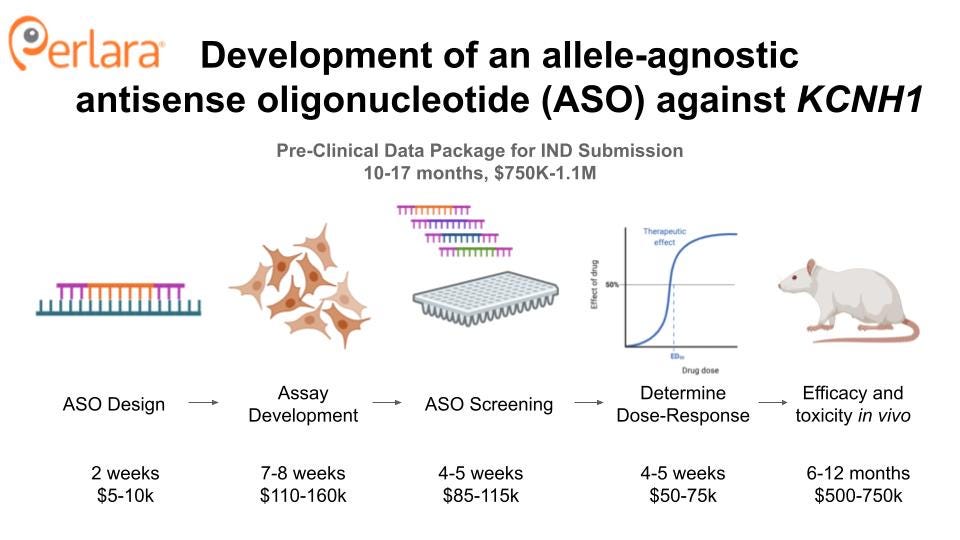
Development of a KCNH1-targeted antisense oligonucleotide
Antisense oligonucleotides (ASOs) are short oligonucleotides that modulate gene expression by complementary base pairing to the mRNA of the targeted gene. ASOs can be employed to promote exon inclusion or exclusion, or to knock down gene expression by targeting mRNAs for degradation. They are an especially attractive therapeutic modality because of their high sequence specificity; for this application, an ASO may differentiate between hEAG1 and hERG channel expression levels whereas a small molecule channel blocker would not. ASO therapies are already FDA-approved in other pediatric neurodevelopmental disorders like Dravet Syndrome and Spinal Muscular Atrophy, providing a precedent for ASO use in rare, pediatric CNS disease.
ASOs can be allele-specific or non-specific (i.e. allele-agnostic). In heterozygous individuals, allele-specific ASOs can be employed to maintain expression of the wild type copy of the gene; however, developing truly allele-specific yet efficient ASOs is challenging and expensive (requiring $1,000,000 per year). Given that knockdown of Kcnh1 is well-tolerated in mice, and the observation that individuals that are heterozygous for a KCNH1 nonsense mutation are not particularly affected, we propose the development of a non-selective ASO as the fastest and most effective way forward. In addition to being easier to develop, the potential patient population able to be enrolled in a clinical trial and ultimately treated will be greater than that for an allele-selective ASO.

The ASO will be designed as a 2-MOE gapmer, and will target a region of the gene that is conserved between mice and and rats and humans so that the same compound can be used throughout the preclinical data package, without the need for a humanized mouse model and offering the ability to assess KCNH1 knockdown, i.e. target engagement, in a rat brain following intrathecal administration.
Pilot experiments will be performed to determine the appropriate cell lines to use for a primary screen (e.g. primary neuronal, patient-derived, immortal) of KCNH1 expression. Candidate ASOs identified by the primary screen will be further characterized for their effect on KCNH1 expression and activity in Tristan’s iPSC differentiated neurons. Mouse models will be used to test for potency and safety, as well as to determine route of administration and therapeutic potential associated with delivery at different stages of development, with zebrafish models considered an alternative strategy if the mouse model fails to recapitulate a relevant phenotype.
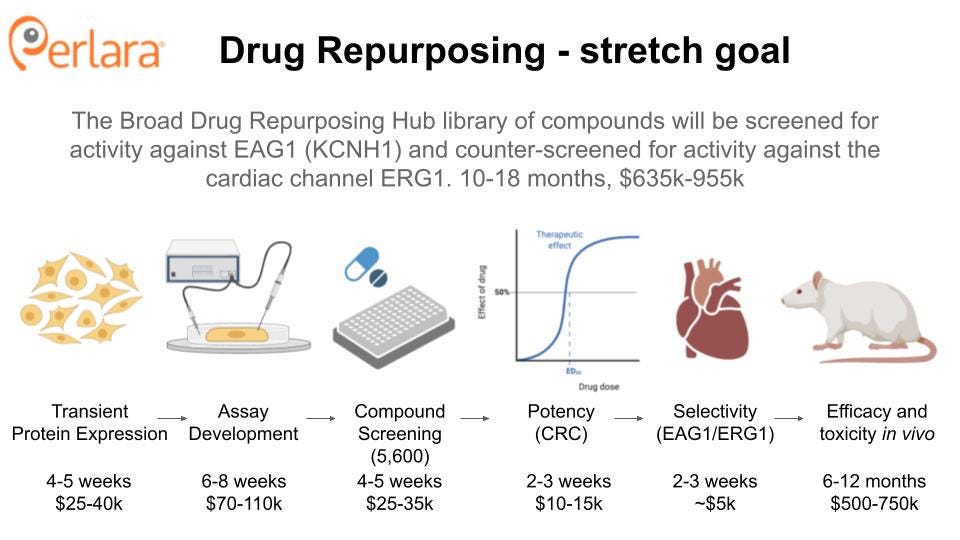
THERAPEUTIC TRACK 2: DRUG REPURPOSING
Drug repurposing is defined as identifying a new use for an already existing drug, be it a century-old drug or a drug that was just recently FDA-approved. Drug repositioning is defined as salvaging an experimental drug that cleared the initial hurdle of early-stage safety studies but failed in late-stage efficacy studies for its intended disease indication.
TRACK 2A: An unbiased drug repurposing screen
Identification of a drug that selectively blocks hEAG1 has been difficult due to its similarity to the cardiac channel hERG1 (Toplak et al. 2022). To address this, we propose an unbiased drug repurposing screen of wild-type and mutant channels to identify compounds that might selectively inhibit hEAG1 (wild type or mutant). The Broad Repurposing Hub (commercially available as the SPECS library) is an ideal launchpad for identifying drug repurposing and drug repositioning candidates. The library includes 4,707 compounds, including 3,422 drugs that are currently marketed or have been tested in clinical trials in humans (Corsello et al. 2017).
It is possible that Tristan’s variant (or other variants) introduce changes into the protein that will make it easier to target than the wild-type protein. It is also possible that a compound that inhibits the wild-type protein will be unable to inhibit the mutant channel. To account for these possibilities, we propose a primary screen of cell lines (e.g. HEK293) expressing recombinant wild-type hEAG1 or Tristan’s variant. hEAG1 activity will be assayed using a fluorescence-based screen (e.g. thalium-flux assay) or electrophysiological readout (Yu et al. 2016). This assay could easily be expanded to include other KCNH1 variants in the future.
Lead compounds will be counter-screened for hERG1 toxicity, as is industry standard and required by the FDA. Compounds that do not show significant inhibition of hERG1 will be further characterized in neuronal cell lines and tested for their ability to restore cilia formation in a recombinant cell line. Finally, drug candidates will be tested for their efficacy and safety in worms and flies and/or a mouse model.
TRACK 2B: Imipramine for seizure control in a small cohort
Imipramine (brand name Tofranil) is a tricyclic antidepressant commonly used to treat mood disorders prior to the development of selective serotonin reuptake inhibitors (SSRIs); its use dramatically decreased after the introduction of SSRIs because of its side effect profile (e.g.. sedation, cognitive slowing, constipation, urinary retention) and its lethality attributable to cardiotoxicity when taken in overdose. Interestingly, because of its anticholinergic effects such as urinary retention, imipramine has been tested at low doses (10-50mg qHS) administered at bedtime in children under 19 with enuresis in at least 14 clinical trials (National Clinical Guideline Centre (UK) 2011). It is no longer commonly used for this purpose, having been replaced by enuresis alarms and intranasal desmopressin, but the data from these trials provides precedent for a pediatric indication, dosage, and safety monitoring in a relevant population.
Imipramine has been demonstrated in vitro to block hEAG1 in a voltage-dependent fashion with hysteresis. Preliminary work on KCNH1-expressing oocytes, including Tristan’s variant, suggests imipramine blocks hEAG1; however, it also blocks the cardiac channel hERG1. Under close monitoring by a cardiologist, it is possible that imipramine could be safely used off-label to “treat” of KCNH1-related disorders. The challenge in this proposal is twofold: clinically-relevant trial endpoints will be essential to both providing quantifiable evidence of efficacy, if present, but also identifying if (and where) the therapeutic window may lie in terms of dosing range.
In other words, it needs to be determined if there is a dose at which imipramine may prevent or shorten seizure frequency and duration, respectively, and/or improve cognitive and motor development. Whereas seizures are not as prominent in KCNH1-related neurodevelopmental diseases as other channelopathies causing epilepsy, EEG biomarkers such as interictal discharges and background disorganization (i.e. excess voltage, delta/theta waves) may be identifiable in an initial test cohort that may also reveal an initial impact (or lack of impact) of low-dose imipramine on CNS function. As such, we propose testing the efficacy of imipramine at controlling seizures in a small cohort of patients. The major benefit to this approach is that the pharmacotherapy is already available and its safety profile is albeit not ideal but well-understood.
BEYOND KCNH1-RELATED NEURODEVELOPMENTAL DISORDERS
Therapeutics developed for KCNH1-related neurodevelopmental disorders have the potential to be applied as treatments for cancer, given the prevalence of ectopic KCNH1 isoforms in neoplasms. This topic is beyond the scope of this roadmap, but may be worthy of further exploration once either an ASO or a small molecule channel blocker has been identified.

FUNDING MODEL
Cure KCNH1, LLC.
ROADMAP UPDATES
This Roadmap is considered version 1 (v1) and it will require annual comprehensive reviews to incorporate progress reports and course corrections.
REFERENCES
Aubert Mucca, Marion, Olivier Patat, Sandra Whalen, Lionel Arnaud, Giulia Barcia, Julien Buratti, Benjamin Cogné, et al. 2021. “Patients with KCNH1-Related Intellectual Disability without Distinctive Features of Zimmermann-Laband/Temple-Baraitser Syndrome.” Journal of Medical Genetics, April. https://doi.org/10.1136/jmedgenet-2020-107511.
Bai, Yifeng, Hongzhan Liao, Tianzhu Liu, Xiangping Zeng, Faman Xiao, Luqiao Luo, Hongbo Guo, and Linlang Guo. 2013. “MiR-296-3p Regulates Cell Growth and Multi-Drug Resistance of Human Glioblastoma by Targeting Ether-à-Go-Go (EAG1).” European Journal of Cancer 49 (3): 710–24.
Bramswig, Nuria C., C. W. Ockeloen, J. C. Czeschik, A. J. van Essen, R. Pfundt, J. Smeitink, Poll-The, B. T., et al. 2015. “‘Splitting versus Lumping’: Temple–Baraitser and Zimmermann–Laband Syndromes.” Human Genetics 134 (10): 1089–97.
Bronk, Peter, Elena A. Kuklin, Srinivas Gorur-Shandilya, Chang Liu, Timothy D. Wiggin, Martha L. Reed, Eve Marder, and Leslie C. Griffith. 2018. “Regulation of Eag by Ca2+/calmodulin Controls Presynaptic Excitability in Drosophila.” Journal of Neurophysiology 119 (5): 1665–80.
Cázares-Ordoñez, V., and L. A. Pardo. 2017. “Kv10.1 Potassium Channel: From the Brain to the Tumors.” Biochemistry and Cell Biology = Biochimie et Biologie Cellulaire 95 (5): 531–36.
Chávez-López, María de Guadalupe, Violeta Zúñiga-García, Blanca Elena Castro-Magdonel, Eunice Vera, Efraín Garrido, Janet Sánchez-Ramos, M. Verónica Ponce-Castañeda, et al. 2020. “Eag1 Gene and Protein Expression in Human Retinoblastoma Tumors and Its Regulation by pRb in HeLa Cells.” Genes 11 (2). https://doi.org/10.3390/genes11020119.
Chen, Jun, Zefeng Xuan, Wenfeng Song, Weili Han, Hao Chen, Yehui Du, Haiyang Xie, Yongchao Zhao, Shusen Zheng, and Penghong Song. 2021. “EAG1 Enhances Hepatocellular Carcinoma Proliferation by Modulating SKP2 and Metastasis through Pseudopod Formation.” Oncogene 40 (1): 163–76.
Chen, Ye, Araceli Sánchez, María E. Rubio, Tobias Kohl, Luis A. Pardo, and Walter Stühmer. 2011. “Functional K(v)10.1 Channels Localize to the Inner Nuclear Membrane.” PloS One 6 (5): e19257.
Corsello, Steven M., Joshua A. Bittker, Zihan Liu, Joshua Gould, Patrick McCarren, Jodi E. Hirschman, Stephen E. Johnston, et al. 2017. “The Drug Repurposing Hub: A next-Generation Drug Library and Information Resource.” Nature Medicine 23 (4): 405–8.
Fang, Ya-Ching, Ssu-Ju Fu, Po-Hao Hsu, Pei-Tzu Chang, Jing-Jia Huang, Yi-Chih Chiu, Yi-Fan Liao, Guey-Mei Jow, Chih-Yung Tang, and Chung-Jiuan Jeng. 2021. “Identification of MKRN1 as a Second E3 Ligase for Eag1 Potassium Channels Reveals Regulation via Differential Degradation.” The Journal of Biological Chemistry 296 (January). https://doi.org/10.1016/j.jbc.2021.100484.
Fukai, Ryoko, Hirotomo Saitsu, Yoshinori Tsurusaki, Yasunari Sakai, Kazuhiro Haginoya, Kazumasa Takahashi, Monika Weisz Hubshman, et al. 2016. “De Novo KCNH1 Mutations in Four Patients with Syndromic Developmental Delay, Hypotonia and Seizures.” Journal of Human Genetics 61 (5): 381–87.
Gabriel, Elke, Anand Ramani, Nazlican Altinisik, and Jay Gopalakrishnan. 2020. “Human Brain Organoids to Decode Mechanisms of Microcephaly.” Frontiers in Cellular Neuroscience 14 (May): 115.
Gavrilova-Ruch, O., K. Schönherr, G. Gessner, R. Schönherr, T. Klapperstück, W. Wohlrab, and S. H. Heinemann. 2002. “Effects of Imipramine on Ion Channels and Proliferation of IGR1 Melanoma Cells.” The Journal of Membrane Biology 188 (2): 137–49.
Gómez-Varela, David, Esther Zwick-Wallasch, Hendrik Knötgen, Araceli Sánchez, Thore Hettmann, Dmitri Ossipov, Rüdiger Weseloh, et al. 2007. “Monoclonal Antibody Blockade of the Human Eag1 Potassium Channel Function Exerts Antitumor Activity.” Cancer Research 67 (15): 7343–49.
Gripp, Karen W., Sarah F. Smithson, Ingrid J. Scurr, Julia Baptista, Anirban Majumdar, Germaine Pierre, Maggie Williams, et al. 2021. “Syndromic Disorders Caused by Gain-of-Function Variants in KCNH1, KCNK4, and KCNN3—a Subgroup of K+ Channelopathies.” European Journal of Human Genetics: EJHG 29 (9): 1384–95.
Haitin, Yoni, Anne E. Carlson, and William N. Zagotta. 2013. “The Structural Mechanism of KCNH-Channel Regulation by the Eag Domain.” Nature 501 (7467): 444–48.
Han, Bo, Kunyan He, Chunlin Cai, Yin Tang, Linli Yang, Stefan H. Heinemann, Toshinori Hoshi, and Shangwei Hou. 2016. “Human EAG Channels Are Directly Modulated by PIP2 as Revealed by Electrophysiological and Optical Interference Investigations.” Scientific Reports 6 (1): 1–13.
Han, Bo, Tursonjan Tokay, Guangming Zhang, Peng Sun, and Shangwei Hou. 2017. “Eag1 K+ Channel: Endogenous Regulation and Functions in Nervous System.” Oxidative Medicine and Cellular Longevity 2017 (March): 7371010.
Hemmerlein, Bernhard, Rüdiger M. Weseloh, Fernanda Mello de Queiroz, Hendrik Knötgen, Araceli Sánchez, María E. Rubio, Sabine Martin, et al. 2006. “Overexpression of Eag1 Potassium Channels in Clinical Tumours.” Molecular Cancer 5 (October): 41.
Hsu, Po-Hao, Yu-Ting Ma, Ya-Ching Fang, Jing-Jia Huang, Yu-Ling Gan, Pei-Tzu Chang, Guey-Mei Jow, Chih-Yung Tang, and Chung-Jiuan Jeng. 2017. “Cullin 7 Mediates Proteasomal and Lysosomal Degradations of Rat Eag1 Potassium Channels.” Scientific Reports 7 (1): 1–15.
Hui, Chen, Zhang Lan, Lin Yue-li, Hong Li-lin, and Huang Li-lin. 2015. “Knockdown of Eag1 Expression by RNA Interference Increases Chemosensitivity to Cisplatin in Ovarian Cancer Cells.” Reproductive Sciences 22 (12): 1618–26.
Kohl, Tobias, Eva Lörinczi, Luis A. Pardo, and Walter Stühmer. 2011. “Rapid Internalization of the Oncogenic K+ Channel K(V)10.1.” PloS One 6 (10): e26329.
Kortüm, Fanny, Viviana Caputo, Christiane K. Bauer, Lorenzo Stella, Andrea Ciolfi, Malik Alawi, Gianfranco Bocchinfuso, et al. 2015. “Mutations in KCNH1 and ATP6V1B2 Cause Zimmermann-Laband Syndrome.” Nature Genetics 47 (6): 661–67.
Li, Zhandong, Ketong Zhu, Xin Gong, Steven Vasilescu, Yu Sun, Kaiqing Hong, Hao Li, Lin Li, and Yaming Shan. 2017. “Inducing Polyclonal Eag1-Specific Antibodies by Vaccination with a Linear Epitope Immunogen and Its Relation to Breast Tumorigenesis.” Pathology Oncology Research: POR 23 (4): 761–67.
Ma, Linlin, Yanni K.Y. Chin, Zoltan Dekan, Volker Herzig, Chun Yuen Chow, Jacqueline Heighway, Sau Wing Lam, Gilles J. Guillemin, Paul F. Alewood, Glenn F. King. 2018. “Novel venom-derived inhibitors of the human EAG channel, a putative antiepileptic drug target.” Biochemical Pharmacology 158: 60-72.
Mastrangelo, Mario, Ingrid E. Scheffer, Nuria C. Bramswig, Lal D. V. Nair, Candace T. Myers, Maria Lisa Dentici, Georg C. Korenke, et al. 2016. “Epilepsy in KCNH1-Related Syndromes.” Epileptic Disorders: International Epilepsy Journal with Videotape 18 (2): 123–36.
Mégarbané, André, Rashid Al-Ali, Nancy Choucair, Monko Lek, Ena Wang, Moncef Ladjimi, Catherine M. Rose, et al. 2016. “Temple-Baraitser Syndrome and Zimmermann-Laband Syndrome: One Clinical Entity?” BMC Medical Genetics 17 (1): 42.
Mello de Queiroz, Fernanda, Guilherme Suarez-Kurtz, Walter Stühmer, and Luis A. Pardo. 2006. “Ether à Go-Go Potassium Channel Expression in Soft Tissue Sarcoma Patients.” Molecular Cancer 5 (October): 42.
Meyer, R., R. Schönherr, O. Gavrilova-Ruch, W. Wohlrab, and S. H. Heinemann. 1999. “Identification of Ether à Go-Go and Calcium-Activated Potassium Channels in Human Melanoma Cells.” The Journal of Membrane Biology 171 (2): 107–15.
Napp, Joanna, Francisco Monje, Walter Stühmer, and Luis A. Pardo. 2005. “Glycosylation of Eag1 (Kv10.1) Potassium Channels: Intracellular Trafficking and Functional Consequences.” The Journal of Biological Chemistry 280 (33): 29506–12.
National Clinical Guideline Centre (UK). 2011. Nocturnal Enuresis: The Management of Bedwetting in Children and Young People. London: Royal College of Physicians (UK).
Niday, Zachary, and Anastasios V. Tzingounis. 2018. “Potassium Channel Gain of Function in Epilepsy: An Unresolved Paradox.” The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry 24 (4): 368–80.
Ouadid-Ahidouch, Halima, Ahmed Ahidouch, and Luis A. Pardo. 2016. “Kv10.1 K(+) Channel: From Physiology to Cancer.” Pflugers Archiv: European Journal of Physiology 468 (5): 751–62.
Pardo, L. A., D. del Camino, A. Sánchez, F. Alves, A. Brüggemann, S. Beckh, and W. Stühmer. 1999. “Oncogenic Potential of EAG K(+) Channels.” The EMBO Journal 18 (20): 5540–47.
Pardo, Luis A., and Walter Stühmer. 2008. “Eag1: An Emerging Oncological Target.” Cancer Research 68 (6): 1611–13.
Sánchez, Araceli, Diana Urrego, and Luis A. Pardo. 2016. “Cyclic Expression of the Voltage-Gated Potassium Channel KV10.1 Promotes Disassembly of the Primary Cilium.” EMBO Reports 17 (5): 708–23.
Schönherr, R., K. Löber, and S. H. Heinemann. 2000. “Inhibition of Human Ether à Go-Go Potassium Channels by Ca(2+)/calmodulin.” The EMBO Journal 19 (13): 3263–71.
Simons, Cas, Lachlan D. Rash, Joanna Crawford, Linlin Ma, Ben Cristofori-Armstrong, David Miller, Kelin Ru, et al. 2015. “Mutations in the Voltage-Gated Potassium Channel Gene KCNH1 Cause Temple-Baraitser Syndrome and Epilepsy.” Nature Genetics 47 (1): 73–77.
Stengel, Rayk, Eric Rivera-Milla, Nirakar Sahoo, Christina Ebert, Frank Bollig, Stefan H. Heinemann, Roland Schönherr, and Christoph Englert. 2012. “Kcnh1 Voltage-Gated Potassium Channels Are Essential for Early Zebrafish Development.” The Journal of Biological Chemistry 287 (42): 35565–75.
Toplak, Žan, Louise A. Hendrickx, Reham Abdelaziz, Xiaoyi Shi, Steve Peigneur, Tihomir Tomašič, Jan Tytgat, Lucija Peterlin-Mašič, and Luis A. Pardo. 2022. “Overcoming Challenges of HERG Potassium Channel Liability through Rational Design: Eag1 Inhibitors for Cancer Treatment.” Medicinal Research Reviews 42 (1): 183–226.
Toplak, Žan, Franci Merzel, Luis A. Pardo, Lucija Peterlin Mašič, and Tihomir Tomašič. 2021. “Molecular Dynamics-Derived Pharmacophore Model Explaining the Nonselective Aspect of KV10.1 Pore Blockers.” International Journal of Molecular Sciences 22 (16). https://doi.org/10.3390/ijms22168999.
Ufartes, Roser, Tomasz Schneider, Lena Sünke Mortensen, Camino de Juan Romero, Klaus Hentrich, Hendrik Knoetgen, Vadim Beilinson, et al. 2013. “Behavioural and Functional Characterization of Kv10.1 (Eag1) Knockout Mice.” Human Molecular Genetics 22 (11): 2247–62.
Urrego, Diana, Naira Movsisyan, Roser Ufartes, and Luis A. Pardo. 2016. “Periodic Expression of Kv10.1 Driven by pRb/E2F1 Contributes to G2/M Progression of Cancer and Non-Transformed Cells.” Cell Cycle 15 (6): 799–811.
Wang, Hui, Xiaohua Zhang, and Hongfang Ding. 2021. “Temple-Baraitser Syndrome with KCNH1 Asn510Thr: A New Case Report.” Clinical Dysmorphology 30 (1): 27–31.
Wang, Xuzhao, Yafei Chen, Junwei Li, Shuai Guo, Xiaoe Lin, Hailin Zhang, Yong Zhan, and Hailong An. 2019. “Tetrandrine, a Novel Inhibitor of Ether-à-Go-Go-1 (Eag1), Targeted to Cervical Cancer Development.” Journal of Cellular Physiology 234 (5): 7161–73.
Wang, Xuzhao, Yafei Chen, Yuhong Zhang, Shuai Guo, Li Mo, Hailong An, and Yong Zhan. 2017. “Eag1 Voltage-Dependent Potassium Channels: Structure, Electrophysiological Characteristics, and Function in Cancer.” The Journal of Membrane Biology 250 (2): 123–32.
Wang, Ze-Jun, Stephanie M. Soohoo, Purushottam B. Tiwari, Grzegorz Piszczek, and Tinatin I. Brelidze. 2020. “Chlorpromazine Binding to the PAS Domains Uncovers the Effect of Ligand Modulation on EAG Channel Activity.” The Journal of Biological Chemistry 295 (13): 4114–23.
Weber, Claudia, Fernanda Mello de Queiroz, Bryan R. Downie, Arnt Suckow, Walter Stühmer, and Luis A. Pardo. 2006. “Silencing the Activity and Proliferative Properties of the Human EagI Potassium Channel by RNA Interference.” The Journal of Biological Chemistry 281 (19): 13030–37.
Whicher, Jonathan R., and Roderick MacKinnon. 2016. “Structure of the Voltage-Gated K+ Channel Eag1 Reveals an Alternative Voltage Sensing Mechanism.” Science 353 (6300): 664–69.
Wrede, Randi von, Monika Jeub, Idil Ariöz, Christian E. Elger, Hubertus von Voss, Hanns-Georg Klein, Albert J. Becker, Susanne Schoch, Rainer Surges, and Wolfram S. Kunz. 2021. “Novel KCNH1 Mutations Associated with Epilepsy: Broadening the Phenotypic Spectrum of KCNH1-Associated Diseases.” Genes 12 (2). https://doi.org/10.3390/genes12020132.
Wu, Jin, Xinyu Wu, Daixing Zhong, Wenliang Zhai, Zhenqi Ding, and Yong Zhou. 2012. “Short Hairpin RNA (shRNA) Ether à Go-Go 1 (Eag1) Inhibition of Human Osteosarcoma Angiogenesis via VEGF/PI3K/AKT Signaling.” International Journal of Molecular Sciences 13 (10): 12573–83.
Wulff, Heike, Neil A. Castle, and Luis A. Pardo. 2009. “Voltage-Gated Potassium Channels as Therapeutic Targets.” Nature Reviews. Drug Discovery 8 (12): 982–1001.
Yu, Hai-Bo, Min Li, Wei-Ping Wang, and Xiao-Liang Wang. 2016. “High Throughput Screening Technologies for Ion Channels.” Acta Pharmacologica Sinica 37 (1): 34–43.
Zhang, Jiao, Yau-Chi Chan, Jenny Chung-Yee Ho, Chung-Wah Siu, Qizhou Lian, and Hung-Fat Tse. 2012. “Regulation of Cell Proliferation of Human Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells via Ether-à-Go-Go 1 (hEAG1) Potassium Channel.” American Journal of Physiology. Cell Physiology 303 (2): C115–25.
Zhang, Ying-Ying, Jianbo Yue, Hui Che, Hai-Ying Sun, Hung-Fat Tse, and Gui-Rong Li. 2014. “BKCa and hEag1 Channels Regulate Cell Proliferation and Differentiation in Human Bone Marrow-Derived Mesenchymal Stem Cells.” Journal of Cellular Physiology 229 (2): 202–12.