

MCTO Cure Roadmap
The second family to engage us on a Cure Roadmap project is the driving force behind a cure-focused foundation called Sophie's Neighborhood based in Boulder Colorado.


Multicentric Carpotarsal Osteolysis (MCTO) Cure Roadmap
Prepared for Sophie’s Neighborhood by Perlara PBC
December 2021

VISION
Toward a future where people with MCTO live healthy and complete lives. This MCTO Cure Roadmap aims to leverage the existing resources, disease knowledge, and strong working partnerships between Sophie’s Neighborhood and academic and biotech groups to develop a timely, ambitious but de-risked, multi-modality pathway to the first approved MCTO medicine. Given the absence of any therapeutic approach that reverses or even significantly delays the development of severe and life-altering skeletal abnormalities in MCTO, and the nearly universal penetrance of skeletal symptoms compared to the incomplete penetrance of MCTO-related renal disease, this roadmap will focus on ameliorating the skeletal disease burden in MCTO. Some therapeutic tracks could yield agents which act on both the skeletal and renal aspects of MCTO. Sizable overlaps between the targets of the affected transcription factor MAFB and common diseases in a diversity of tissue and organ systems, as well as likely overlaps with affected pathways in other rare genetic diseases, suggests that a drug development program for MCTO could improve the lives of many patients worldwide.
This roadmap proposes in Year 1 to focus on:
initiating and completing a drug repurposing screen,
fully exploring MCTO candidate therapeutics, bone marrow transplantation (BMT) and a potential potent MAFB inhibitor, 3′-sialyllactose
In parallel, the basic science underlying MCTO will be strengthened through:
genetic identification of the MCTO skeletal effector cell type(s),
thoroughly establishing the kinetics of MCTO MAFB variant perdurance in renal and skeletal cell types,
quickly determining whether senescence is a contributing factor in MCTO pathogenesis, and
further establishing the natural history of MCTO.
Finally, back-up therapeutic tracks of
MAFB antisense oligonucleotide (ASO)
MAFB targeted protein degrader, and
mining the existing natural human variation among a MAFB affected and unaffected carrier family members to identify biologically safe, existing modifiers of MCTO pathogenesis are considered and discussed.
AUTHORS
Martha Klovstad, PhD (Perlara Cure Guide)
Michelle Dookwah-Smith, PhD (Perlara Cure Guide)
Ethan O. Perlstein, PhD (CEO of Perlara PBC & Maggie’s Pearl LLC)
REVIEWERS
Lauren Rosenberg (Sophie’s Neighborhood)
Sophie’s Neighborhood Scientific Advisory Board members
EXECUTIVE SUMMARY
The MCTO Cure Roadmap lays out a path for a timely, ambitious but de-risked multi-modality pathway for MCTO therapeutic development that balances the urgency of patient-led drug development with the need for a drug development plan that understands the resources available to ultra-rare disease communities. As such, this plan proposes to leverage learnings and resources from other diseases and to prioritize development of therapies that have been explored in other disease indications. As MCTO therapeutic candidates are developed, this drug development plan envisions that Sophie’s Neighborhood will proactively seek out partnerships to develop and assess selected candidates in larger disease populations.
In Year 1, this roadmap proposes a drug repurposing screen in a primary skeletal cell type overexpressing a MCTO MAFB variant using the Arpeggio Bio nascent RNA sequencing platform as a readout. In addition, the potential of bone marrow transplantation (BMT) and 3′-sialyllactose, a human milk oligosaccharide (HMO) identified as a potential potent MAFB inhibitor in the Arpeggio Bio databases, as MCTO therapeutics will be fully explored.
In parallel in Year 1, the following four capacity-building activities will occur. First, the basic science underlying MCTO will be strengthened through genetic identification of the MCTO skeletal effector cell type(s) in mice. Second, determining the kinetics of MCTO MAFB variant perdurance in renal and skeletal cell types to solidify working hypotheses and establish the range of MAFB perdurance of known MAFB pathogenic variants. Third, determining whether senescence is a contributing factor in MCTO pathogenesis in order to unlock senolytics/senomorphics as a potential candidate therapeutic pathway. Fourth, further expand knowledge of the natural history of MCTO through a formal natural history study in preparation for clinical studies. Finally, back-up therapeutic tracks of MAFB antisense oligonucleotide (ASO), MAFB targeted protein degrader, and mining the existing natural human variation among affected and unaffected MCTO carrier family members are considered and discussed.
These Year 1 activities should lay the foundation for 3-5 “pioneer” trials (single-patient INDs) in MCTO, followed by the launch of a pivotal trial in 10-15 MCTO patients within five years. The anticipated natural history study and learnings from other diseases mentioned in this roadmap will lay the foundation for productive pre-IND interactions with the FDA to discuss clinical trial design for a MCTO therapeutic. If the therapeutic candidate has indication crossover potential, partnership with other organizations will allow for safety assessment in larger populations. Sophie’s Neighborhood will likely be in the best position to drive MCTO therapeutic development either as a co-developer/co-owner with an established industry partner, or as a licensor of specific therapeutic assets to different industry partners or investor syndicates.
INTRODUCTION
What is Multicentric Carpal Tarsal Osteolysis (MCTO)?
Multicentric Carpal Tarsal Osteolysis, or MCTO, is an ultra-rare skeletal dysplasia characterized by progressive osteolysis of the carpal and tarsal bones. A recent review by Wu et al. (2021) assessed the 51 currently published cases of MCTO. A graph summarizing the pathological features of the patients is seen in Figure 1. All patients presenting with MCTO experience the progressive bone lesions in the hands and feet. While the larger bone joints of the knees and elbows are also frequently involved, there are few reports of hip, shoulder, or spinal involvement - of the 51 patients summarized in this review, four of them have scoliosis. Other symptoms seen in some patients include craniofacial abnormalities such as triangular face shape, micrognathia (small lower jaw), and exophthalmos (bulging eyes). Other eye disorders have also been reported including clouding of the cornea. Two-thirds of the patients report renal lesions, with half of those patients experiencing renal failure. The renal disease often first presents as proteinuria and is pathologically characterized as nephropathy with focal segmental glomerulosclerosis (FSGS).
Figure 1: MCTO Pathology, Percent of Patients Affected by Pathological Feature

Source: Wu, 2021
N = 51 (bone lesions), 31 (Non-wrist/ankle joint involvement), 48 (renal lesions), 32 (renal failure), 17 (eye abnormalities), 13 (facial abnormalities), 15 (other manifestations including ENT, lymphoma, respiratory, epilepsy, Chiari I malformation, cardiology, learning disability), 49 (family history)
Wu et al (2021) provides an overview of the timeline by which pathological phenotypes typically occurred within the 51 published cases of MCTO, which can be seen in Table 1. Over half of the cases (N=31) reported skeletal symptom onset by two years of age. However, the median age of diagnosis is 11 years of age and the median delay in diagnosis was 3.83 years. Due to the presentation of osteolysis prior to other MCTO symptoms, patients are often misdiagnosed as having juvenile rheumatoid arthritis. Delayed diagnosis results in delayed and ineffective symptom management. Progressive destruction of the carpal and tarsal bones occurs in all MCTO patients, and deformities of the knee and elbow joints are also present but are not necessarily symmetrical. These lesions result in joint pain and loss of mobility caused by locked elbows and knees, which can continue to progress and result in severe functional impairment. For patients with severe bone damage, surgical treatment is required.
Renal dysfunction typically presents after the onset of skeletal symptoms, with the median age of onset of renal lesions occurring at almost six years of age (N=21). However, half of the patients who presented with renal lesions progressed to end-stage renal disease, with the median time frame of three years from initial identification of renal lesions to renal failure. For the two-thirds of patients with known renal lesions (32/48), traditional oral steroids and/or immunosuppressive drugs were not effective. However, there was a single case where the patient was treated successfully with cyclosporine A (Kaimori, 2018). ACE inhibitors can be utilized to treat proteinuria in patients; however, half of the patients still progressed to renal failure, at which point dialysis and kidney transplants are required. Patients who have received a kidney transplant have not shown relapse of renal lesions following transplantation (Matthew Sampson, personal communication).
Table 1: Age of Onset of MCTO Pathologies and Other Disease Characteristics
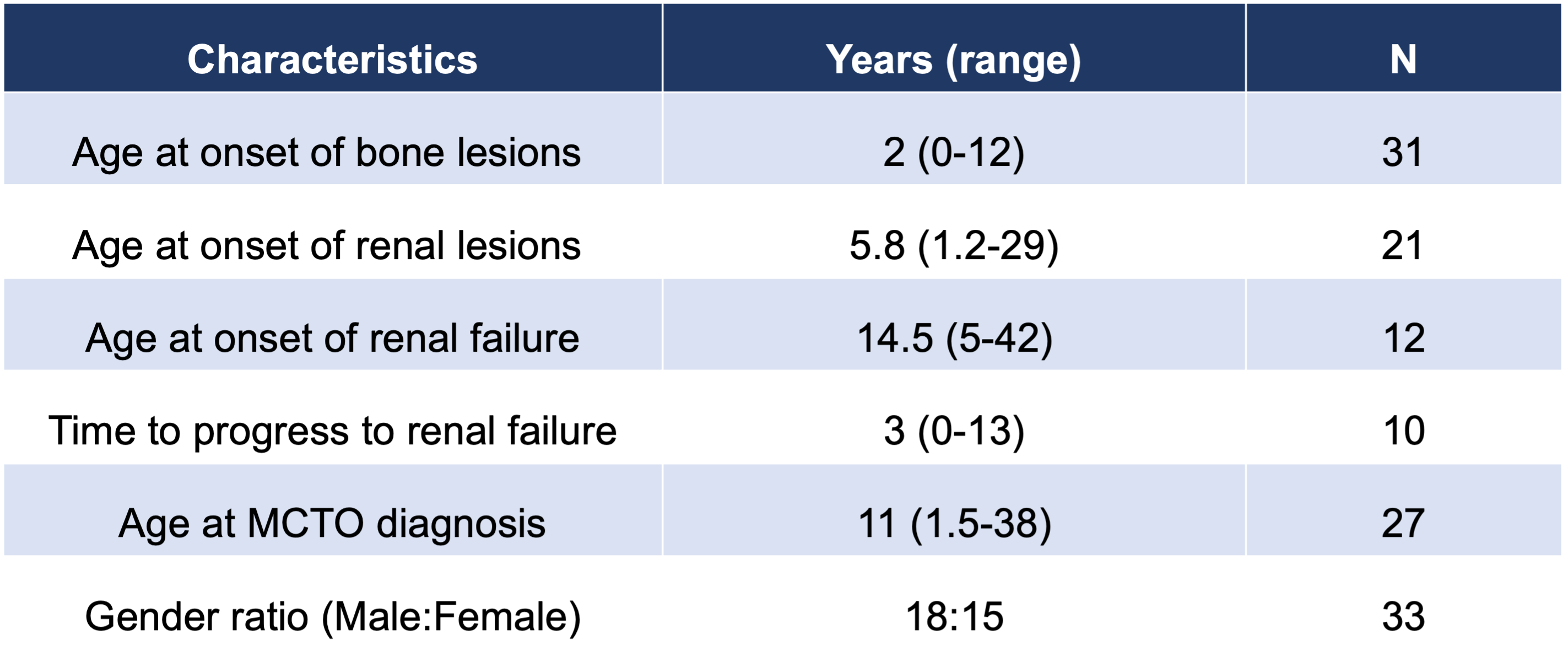
Source: Wu, 2021
MCTO is a clinically heterogeneous disorder with patients experiencing varying phenotypes in different organ systems, with differences in disease onset and rate of disease progression from patient to patient. Wu et al also investigated whether or not genotype contributed to the clinical heterogeneity in MCTO. However, their assessment found no clear links between specific gene mutations and the clinical phenotypes. Additional cases and a better understanding of disease mechanisms may help elucidate a connection between genotypes and clinical manifestations of MCTO.
MCTO is caused by heterozygous missense mutations within an approximately 50 stretch of the N-terminal transactivation domain of MAFB, a basic leucine zipper (bZIP) transcription factor that is involved in multiple biological processes. MAFB is expressed in many tissues. Of note for MCTO, MAFB is expressed selectively in monocytes and macrophages but not in other hematopoietic lineages, which is distinct from many other myeloid transcription factors. MAFB is also expressed in chondrocytes. MAFB roles include controlling lens development (Reza, 2007), lymphangiogenesis (Dieterich, 2015, Dieterich, 2020), pancreatic α and β cell differentiation (Conrad, 2016, Hang, 2011), skin cell differentiation (Lopez-Pajares, 2015), chondrocyte matrix formation and development (Zhang, 2013), and podocyte generation (Moriguchi, 2006, Morito, 2014, Kann, 2015, Dong, 2015), and also regulates type I IFN production through recruitment of coactivators to IFN regulatory factor 3 (Kim, 2010).
MAFB belongs to the MAF family of transcription factors, and is one of the four large MAF transcription factors. MAFB binds Maf Recognition Elements (MAREs) or MARE half-sites. MAF proteins form homo and hetero-dimers via an extended leucine zipper; MAFB can form dimers with many other Activating Proteint-1 (AP1) TF members (MAFA, MAFB, c-MAF, FOS, FOSB, FRA1, FRA2, JUN, BACH1).
The mechanism by which mutations in MAFB cause the varied pathological phenotypes seen in MCTO remains unknown, especially with regard to the skeletal phenotype. All known MCTO mutations are heterozygous missense mutations in an approximately 50 amino acid portion of the N-terminal transactivation domain (See Figure 2). The known mutations run a stretch of approximately 20 amino acids, which have been identified to cause MCTO. There are not 1-2 mutations which predominate, although several have been observed more than once. Two-thirds of MCTO cases are sporadic, but familial cases are known (Wu, 2021). There is variable phenotypic expression, even within the same family (Mehawej, 2013) and for the same mutation across families (six cases of 206C>T, Ser69Leu with no correlation in those cases with respect to disease severity, age of onset, or renal involvement; Upadia, 2018). A C-terminal MAFB nonsense mutation (Ser197Ter) and a mutation in the DNA-binding domain have been shown to cause a Duane Retraction Syndrome, or DRS, which is associated with eye movement abnormalities and with FSGS (Kaimori, 2018, Sato, 2018).
Figure 2: MAFB gene with MCTO and DRS-causing mutations

Source: Hamada, 2020
MCTO Therapeutic Goals
MCTO Therapeutic Goals for Sophie
The primary goal for an MCTO therapeutic for Sophie is to reduce or stabilize carpal-tarsal (CT) lesions where the most disease progression is currently being seen on imaging, with reducing or stabilizing non-CT lesions being of second importance and urgency. While some MCTO patients experience severe joint pain and/or osteoporosis, neither of these conditions appear to affect Sophie at this time and the clinical variability of MCTO suggests that Sophie may or may not develop these conditions; as such, these are not priority therapeutic goals at this time. Up to half of MCTO patients experience kidney function failure due to focal segmental glomerulosclerosis (FSGS). While Sophie has exhibited some mild proteinuria, her kidney function is currently relatively normal on an ACE inhibitor. Given that progression into end-stage kidney disease is possible but may not occur or be years away, development of a MCTO therapeutic that positively impacts renal disease aspects while also positively modulating skeletal structure and function is ideal, but skeletal effects should be prioritized when designing screens or selecting drug candidates. However, given the compromised state of the kidney in MCTO, renal toxicity is a primary concern for any drug candidate and should be directly assessed at an early stage of development.
Table 2: Goals for MCTO Therapeutic Development, as prioritized by Lauren Rosenberg

Overall MCTO Therapeutic Goals
The therapeutic goals of Sophie’s family match the overall unmet needs for the general MCTO population: while MCTO renal disease is a major source of mortality risk in MCTO, not all MCTO patients will be affected and these MCTO sequelae can be cured by kidney transplant if a suitable kidney donation is available (Table 2). In contrast, nearly all MCTO patients are affected by skeletal dysplasia, which is the major source of MCTO morbidity impacts, and MCTO standard of care is largely ineffective at halting skeletal disease progression. Targeting carpal-tarsal abnormalities is the most sensible primary target for MCTO therapeutic development given the nearly complete penetrance among affected MCTO patients and huge impacts on quality of life. Given the biological similarities between CT and large joint abnormalities, it is likely but not given that a therapeutic that would improve CT lesion would also positively affect large joint abnormalities. Given that the underlying cause of MCTO-related joint pain observed in a minority of MCTO patients is unknown, we can only speculate whether a therapeutic affecting CT lesions would be expected to also positively modulate MCTO joint pain.
MAFB in MCTO: Critical Research Gaps
Two key knowledge gaps exist for MCTO: the genetic nature of MCTO mutations and the cell type or types in which the MCTO mutant protein affects the skeletal phenotypes, in particular, the carpal-tarsal abnormalities. The lack of certainty about the MCTO effector cell type significantly increases the risk of drug discovery failure and should be a research priority for Sophie’s Neighborhood in 2022. The lack of strong direct evidence determining the genetic nature of the MCTO mutations (i.e., loss of function [LOF] vs. gain of function [GOF], overexpression/ increased activity vs. dominant negative) is important to keep in mind when designing and interpreting all MCTO experiments. However, definitive elucidation of the genetic mechanism of MCTO in all cell types of interest may not be immediately essential considering the strong biologic plausibility of increased MAFB perdurance based on the identity of the amino acids that are associated with MCTO. However, several basic science experiments are suggested below that will significantly de-risk the endeavor by confirming the genetic mechanism and potentially shed light on the underlying affected pathways in MCTO.
Genetic Nature of MCTO Mutations
The working hypothesis among several of Sophie’s Neighborhood disease expert scientific advisory board (SAB) members is that MCTO mutations inhibit the phosphorylation of MCTO, leading to increased protein stability and a resulting overexpression-like phenotype. The strength of this hypothesis was confirmed by MafA/B expert Roland Stein (Vanderbilt) based on the identity of mutations causing MCTO. However, the view of MCTO mutations as a GOF, overexpression phenotype is not universally held among Sophie’s Neighborhood SAB members and a few members think that the mutational mechanism of MCTO may be tissue-specific, based on the renal MCTO phenotypes resembling LOF phenotypes in the kidney.
In support of a GOF mechanism, among the 11 amino acids mutated in MCTO, 6 are known or predicted Ser/Thr phosphorylation sites (Thr58, Ser61, Thr62, Ser66, Ser69, Ser70), three are priming sites for the Ser/Thr protein kinases GSK3 and JNK (Pro59, Pro63, Pro71) and two are predicted to be GSK3 priming sites (Sophie’s Neighborhood, personal communication). These post-translational modification events are predicted to result in increased protein stability, a hypothesis supported by the increased MAFB levels in murine monocytes and M-MØ macrophages mutant for MCTO (Cuevas, 2017; see Figure 3).
Most compellingly, two independent transcriptional analyses in mouse macrophages (Cuevas, 2017) and osteoclasts (Takahashi, ASBMR 2021), showed that MafB KO and MAFB MCTO mutant mice display opposite transcriptional profiles. Taken together with additional evidence (Table 3), these results strongly suggest that, at least in the macrophage lineage, the MAFB MCTO mutation is a GOF, overexpression-like mutation. Notably, the MCTO mutation is sometimes described as a dominant negative mutation (meaning that the mutant MAFB protein interferes with the function of the wild-type MAFB protein); currently there is no evidence for a dominant negative mechanism for MCTO mutations.
Table 3: Evidence Supporting a GOF Phenotype for MAFB MCTO Mutations

* Takahashi, results presented at ASBMR 2021; ** Takahashi, personal communication, data not available for review
Figure 3: MAFB perdurance in MCTO monocyte, M-MØ macrophages, and after HEK293T transfection

(C/D) Patient-derived Ser54Leu MCTO monocytes(C) and M-Mφ macrophages (D) (MCTO #1) have increased MAFB protein levels
(E) Increased MAFB expression/persistence of 3 MCTO variants (Ser54Leu, Pro63Arg, Pro71Ser) + WT control after HEK293T transfection
(F) Decreased transactivation of 2-3 MCTO mutants in MARE-luciferase assay
Source: Cuevas, 2017
However, while a GOF, overexpression mechanism for MCTO mutations is the most compelling current hypothesis, it is important to keep in mind when designing future experiments, that even in the skeletal system, not all MCTO results are consistent with a GOF mechanism (Table 4). Namely, MCTO mutant MAFB mutants exhibited reduced transactivation activity in a MARE-luciferase assay, and WT but not MCTO mafB mRNA injected at the 1-cell stage in zebrafish rescues osteoclast over-proliferation of mafbb KO zebrafish (Table 4). While these experiments may be able to be discounted by MAFB autoregulation and peculiarities of the zebrafish, there is some risk of incorrectly assuming an overexpression phenotype, particularly in non-macrophage lineage cells, so overexpression should eventually be explicitly confirmed in the cell types of interest during therapeutic development.
Table 4: Evidence Supporting a LOF Phenotype for MAFB MCTO Mutations
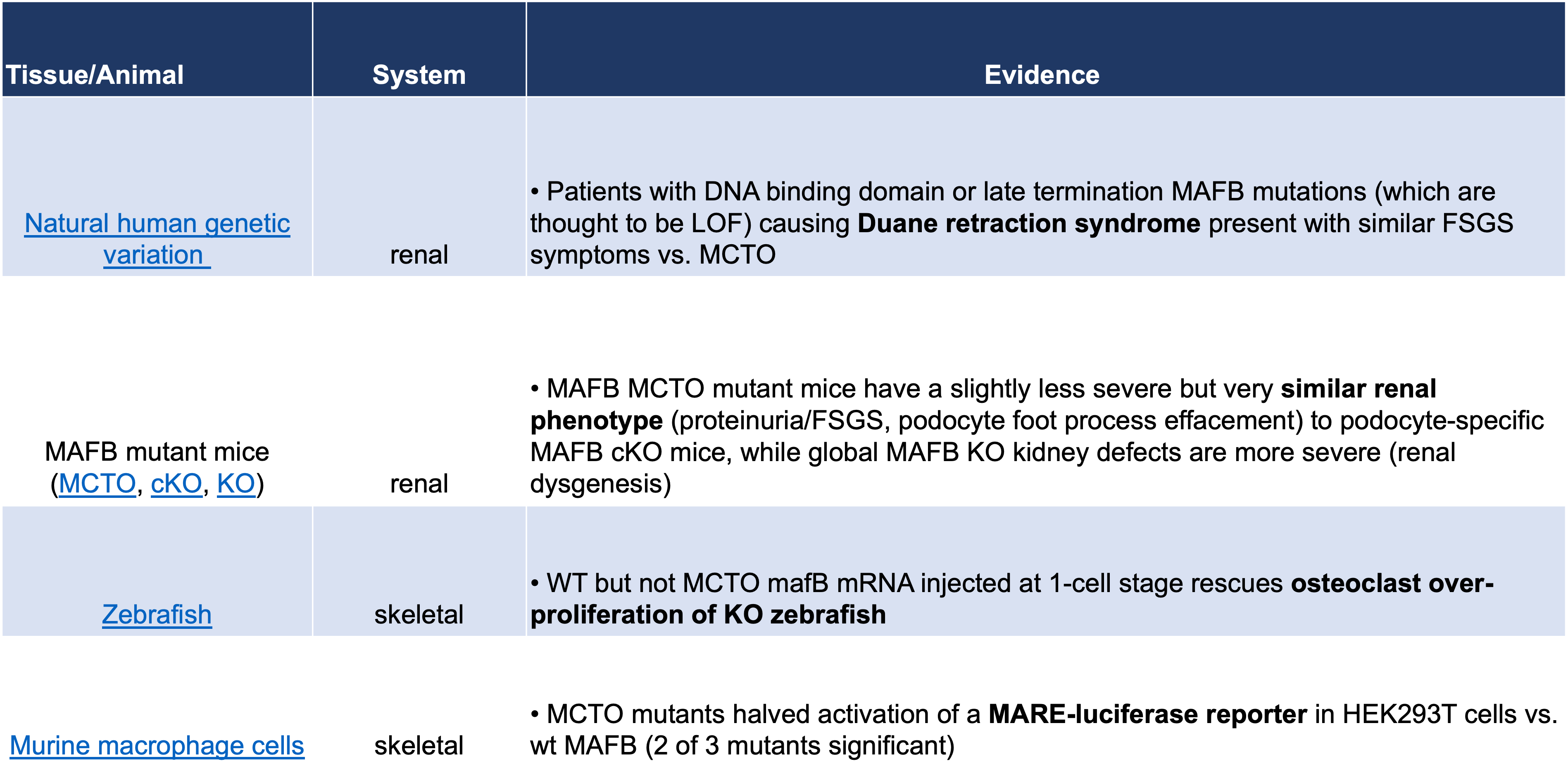
In this vein, the evidence for a GOF mechanism for MCTO is less compelling in the renal system (Tables 3 and 4). MAFB MCTO mutant mice have a slightly less severe but very similar renal phenotype (proteinuria/FSGS, podocyte foot process effacement) to podocyte-specific MAFB cKO mice, while global MAFB KO kidney defects are more severe (renal dysgenesis), suggesting similar defects. In addition, patients with DNA binding domain or late termination MAFB mutations causing Duane retraction syndrome (which are thought to be LOF) present with similar FSGS symptoms as MCTO patients. Therefore to invoke a GOF mechanism for MCTO in the kidney, one would need to invoke a “goldilocks” scenario in which too much or too little MAFB leads to renal dysfunction with the same podocyte foot process effacement phenotype. In support of the idea of too much or too little MAFB causing FSGS, Dr. Saturo Takahashi has unpublished results showing that the MCTO mutant (Pro59Leu) MAFB transcriptional profile is more consistent with MAFB perdurance vs. LOF (personal communication).
In summary, the most likely scenario is a GOF, overperdurance-related phenotype in all tissues. Transcriptional analysis in skeletal effector cells as they are identified and publication of the renal transcriptional analysis in the Pro59Leu MCTO mice would solidify this working model of MCTO.
In addition, two key basic science experiments would de-risk MCTO therapeutic development and provide a more attractive data package for potential partners: demonstration of the perdurance of MAFB protein carrying different MAFB mutations and CHiP-Seq demonstrating the perdurance of MAFB on promoters. MAFB protein perdurance kinetics can be assessed in the skeletal MCTO cell lines being developed by Artisan Bio. In addition, transfection of a larger collection of MAFB variants known to cause MCTO into a more generally available cell line could be a powerful demonstration that perdurance of MAFB is the defining feature of MCTO, as well as determining the kinetic range among different MAFB variants, knowledge that would be critical if a ASO therapeutic path is selected and useful for any therapeutic path. Finally, CHiP-Seq experiments will demonstrate the MCTO MAFB variants persist on key promoters and could identify novel affected pathways if the experiments are done in relevant cell types. If Therapeutic Track 1 is pursued with Arpeggio, initial proof-of-concept experiments may collect the similar data to CHiP-Seq and CHiP-Seq may be unnecessary.
Identity of MCTO Skeletal Effector Cells
Elucidating the effector cell types of the MCTO pathology in the skeletal system is considered both essential and urgent for further MCTO drug development. Due to the osteolytic phenotype of MCTO, osteoclasts were first considered as the candidate MCTO skeletal effector cell. However, antiresorptive therapies, such as the RANKL inhibitor denosumab and bisphosphonates, do not reverse or prevent disease progression, and a more generalized osteolytic phenotype throughout the skeletal system might be expected if MCTO MAFB variants were acting to generally activate osteoclast activity. Moreover, x-rays from Sophie and other young MCTO patients with published x-rays show that there is some element of inappropriate bone development, as viso-spatially the affected joints are smaller and narrower compared to unaffected children, even if there is no bone formation yet. Specifically, Sophie’s wrist cartilaginous joint space is much narrower compared to unaffected children, suggesting a role for chondrocytes and possibly osteoblasts. There is likely an osteoclast component to MCTO, as some MCTO children experience typical development of wrist ossification centers and then the bones dissolve - but increased osteoclast activity is unlikely to explain key aspects of MCTO pathology. (Nina Ma, personal communication).
Instead, chondrocytes, osteoblasts, or osteocytes may be central to the abnormal development of the carpal-tarsal bones in MCTO patients (all from the osteochondro-progenitor lineage). In line with this hypothesis of the osteochondro-progenitor lineage being key effectors of the MCTO pathology, Lazarus and colleagues (2017) argued that the carpotarsal osteolysis disorders MCTO and MONA (multicentric osteolysis nodulosis and arthropathy) may not be typical osteolytic disorders but instead due defects in subarticular ossification (mineralization of a cartilaginous template of the mature bone structure) mediated by MAFB in chondrocytes based on correlations of MAFB and MMP2 temporal and spatial gene expression patterns with the specificity for the carpal-tarsal bones. Moreover, MAFB has been shown to regulate chondrocyte gene expression and cartilage matrix homeostasis via control of aggrecan, mmp3, and mmp13 expression (Zhang, 2013). A role for MAFB in osteoblast-regulated bone remodeling has also been suggested (Ramirez-Salazar, 2020). Alternatively monocytes or macrophages could be MCTO effector cells.
Figure 4: Key Skeletal Lineages: Macrophage/Monocyte Lineage and Mesenchymal Osteochondral-Progenitor Lineage

Source: Yao, 2021 and Tam, 2018
Candidate MCTO effector cell types reside in the hematopoietic stem cell lineage (macrophages, osteoclasts, monocytes) and the mesenchymal cell lineage (osteoblasts, chondrocytes, and to a lesser extent osteocytes) (Figure 4). Determining which cell lineage and cell types are the MCTO effectors is key to several steps of MCTO therapeutic development. First, any drug not delivered systemically will need to be targeted to effector cells, so knowing the target cell surfaces for therapeutic entry as well as the anatomical location relative to the affected joint is key, particularly if an ASO therapeutic track is pursued.
For a drug repurposing approach, a phenotypic screen is attractive, but such a screen is not feasible without knowing the effector cell type(s). While candidate drug hits can be identified in non-effector cell types expressing MAFB, many of the hits from such a screen would be expected to be specific to the screening cell. For example, screening for MAFB MCTO variant suppressors in a podocyte cell culture is an attractive cell model for drug discovery (see the section on Cell-based models for further details) that was deprioritized for MCTO drug discovery based on the incomplete penetrance of renal disease in MCTO patients and later age of development relative to skeletal abnormalities. Many of the hits in a podocyte screen would be expected to be downstream podocyte-specific transcriptional targets of MAFB, such as Nphs1, Magi2, and Tcf21 (Usui, 2000), which would not be expected to impact skeletal pathology. Even many upstream MAFB modifiers that affect MAFB activity would be expected to be cell specific, as the transcriptional regulatory complexes and trans-acting factors impacting MAFB likely have cell-specific components and it is these cell specific components that we hope to find in a suppressor screen in order to minimize off-target adverse effects in non-skeletal and non-renal tissues.
Finally, having strong evidence of a biological rationale for a drug candidate requires knowing the cell type(s) involved in disease pathology. Given the ultra-rare prevalence of MCTO, MCTO clinical trials by necessity will be small. A strong regulatory package will include compelling preclinical proof-of-concept experiments demonstrating modification of disease phenotypes not only in mice, but also in human cell types known to be central effectors of MCTO pathology.
The definitive experiment to determine the skeletal effector cell would be to knock-in the MAFB P59L/P59L variant tissue specifically in a MAFB KO background (Tsunakawa, 2019, Tran, 2016). Considering that this would be a labor-intensive project and which may not be feasible for Dr. Takahashi’s lab in 2022, a quote has been generated from Cyagen in which a conditional MAFB MCTO mutant knock-in is created in the ROSA26 locus in mafBflox/flox mice (see Appendix; quotes from additional companies could be secured); it may also be possible to insert an IRES and GFP downstream of MafB MCTO in order to easily confirm expression (not included in quote). Alternatively, a simpler conditional overexpression of the MAFB Pro59Leu variant from the ROSA26 locus may be somewhat time saving and could be equally informative given the lack of evidence for a dominant negative mechanism in MCTO.
Suitable Cre-lines could be used to tissue specifically express MAFB MCTO mutant in order to phenocopy skeletal aspects of the MAFB MCTO/MCTO mice and precisely determine the MCTO effector cell type(s) (Tsunakawa, 2019; skeletal defects described at ASBMR 2021). A variety of Cre lines exist for osteoclasts, osteocytes, osteoblasts, and chondrocytes (Dallas, 2018; Chen, 2019; Jackson Laboratory) to drive the MAFB MCTO tissue specifically. Cyagen has estimated that it will cost ~30K and 7 months to generate this knock-in mouse from MAFB KO mouse. Additional time will be required to cross these mice with the Cre drivers and assess phenotypes.
An alternative approach to determining the MCTO skeletal effector cell types would be to perform transcriptional analysis to determine which cell types have transcriptional profiles that are altered in MCTO, as is suggested by the Zankl group (Sophie’s Neighborhood, personal communication) or similar to single-cell RNA sequencing experiments done by other groups in related skeletal systems (Sunkara, 2021; Yoshioka, 2021). However, given that MAFB is known to regulate transcription in many skeletal cell types, it may be difficult to match transcriptional changes to phenotypes in the absence of additional genetic information when many cells are affected, even if spatial information is gathered. Therefore, a genetic approach is strongly recommended.
In contrast, while the MCTO skeletal effector cell type(s) are unknown, there is a reasonable level of certainty that MCTO mutations act locally in the kidney, likely in the podocyte cells. MAFB podocyte-specific knockout mice develop overt proteinuria, have higher BUN and serum creatinine levels, FSGS lesions, renal interstitial damage, and podocyte foot process effacement similar to MAFB MCTO mutant mice (Usui, 2000, Tsunakawa, 2019). Most importantly, while there have been some suggestions in the literature of a role for MAFB in renal macrophages (Pan, 2021), MCTO patients who have received a kidney transplant have not shown relapse of renal lesions post-transplantation (Matthew Sampson, personal communication), which largely rules out circulating inflammatory cells as effectors of MCTO renal disease.
THERAPEUTICS READINESS
The development of new medicines requires disease models, patient-derived biosamples and cells, and a deep mechanistic understanding of pathophysiology that identifies disease modifying drug targets. Below is a description of the status of MCTO disease models.
Mouse model
In mice, the same 4 large MAF transcription factors have been identified as in humans - MAFA, MAFB, c-MAF, and NRL. MAFA-, MAFB-, and c-MAF-deficient mice have been generated and their phenotypes have been analyzed (Takahashi, 2021). While MafB−/− neonates die within 24 h after birth (Blanchi, 2003), assessment of the newborn mice has determined that MAFB is essential for the development of pancreatic endocrine cells, formation of inner ears, podocyte function in the kidneys, and functional differentiation of macrophages (Artner, 2007, Cordes, 1994, Moriguchi, 2006, Aziz, 2006). Conditional knockout of MafB in mice found that MAFB is important for the formation of atherosclerotic lesions because it regulates the expression of apoptosis inhibitor of macrophage (AIM), which regulates apoptosis in macrophages (Hamada, 2014). The conditional MafB mouse model was also used to determine that MAFB is required for the maintenance of glomerular podocytes in adult mice (Usui, 2020).
A mouse model with a human MCTO-causing mutation in MAFB was developed by the Takahashi laboratory (Tsunakawa, 2019). They utilized C57BL/6J and Jcl:CD1 (ICR) mice that were obtained from Charles River Laboratories Japan (Kanagawa, Japan) and CLEA Japan (Tokyo, Japan), respectively. The human c.176C>T mutation in the Mafb gene was introduced using the CRISPR/Cas9 technology (Tsunakawa, 2019).
The c.176C>T mutation in the Mafb has been reported to cause MCTO osteolysis and nephropathy in human patients, with pathological phenotypes appearing during early childhood (Zankl, 2012, Mumm, 2014, Mehawej, 2013). The Takahashi lab produced both heterozygous (MafbMCTO/WT) and homozygous (MafbMCTO/MCTO) mice with the c.176C>T (p.Pro59Leu) mutation. The mutant mice exhibited decreased body size relative to the WT mice (MafbWT/WT) mice. Additionally, the mice phenocopy the human patients with regard to renal dysfunction. However, it was determined that the MafbMCTO/MCTO showed a more severe phenotype relative to the heterozygous mice. The homozygous mice exhibited body weight that was 40% lower than that of both MafbWT/WT or MafbMCTO/WT by 4 weeks of age, which continued to be lower at 38 weeks of age. With regard to renal function, the MafbMCTO/MCTO mice exhibited a urine albumin creatinine ratio (ACR) 6 times higher than those of MafbWT/WT at 4 weeks of age, which continued until 38 weeks of age (Tsunakawa, 2019).
The Takahashi laboratory also has recent, unpublished data utilizing their MCTO mouse model investigating skeletal phenotypes relevant to the disease. Consistent with the renal phenotype observed, the MafbMCTO/MCTO mouse exhibits a more severe skeletal phenotype relative to the MafbMCTO/WT mice. Both sets of mice show differences and abnormalities in carpal bones, metacarpal bones, femur bones, and tarsal bones. The mutant mice exhibit decreased body weight and bone volume in all bones relative to wild-type; however, significant differences in the joint bones of MafbMCTO/MCTO relative to MafbMCTO/WT begins at 2 weeks of age. The differences progress through 8 weeks and 16 weeks of age, at which point the homozygous MafbMCTO/MCTO mice display severe skeletal defects, including loss of entire cancellous bone.
An MCTO mouse model with the c.212 C>G; p.Pro71Arg (CCC>CGC) mutation that Sophie carries is in development through the Jackson Laboratory’s Rare and Orphan Disease group. A mouse model with Sophie’s specific pathological mutation can be exceptionally valuable to validate findings and assess potential therapeutics from cell-based experiments in a living animal. This mouse model could also be utilized for evaluating efficacy and dosing for repurposed or novel drugs.
Additionally, another MCTO mouse model is also in development in collaboration with Regeneron Pharmaceuticals. This MCTO mouse model will be a Mafb knockout mouse with a reporter knocked-in to allow for a better understanding of MAFB expression pattern in tissues throughout the whole organism. A reporter mouse line can also be valuable for the evaluation of potential gene therapies for the disease.
Cell-based models
Cell-based disease models are an efficient and cost-effective tool for many applications, from initial basic disease mechanism queries to high-throughput drug screening.
However, a critical part to effectively utilizing cell-based disease models is selecting the appropriate cell type. Determining which cell lineage and cell types are the MCTO effectors is key to several steps of MCTO therapeutic development.
For some experiments, Sophie’s Neighborhood has partnered with Somalogics and Arpeggio Bio to perform proteomic, transcriptomic, and preliminary drug repurposing experiments on Jurkat cells transfected with constitutively expressed wild-type or mutant MAFB. The mutant cell lines are transfected with plasmids expressing either the mutation that Sophie carries, p.Pro71Arg (CCC>CGC): c.212 C>G, or the mutation currently being used in the aforementioned mouse model used by the Takahashi Laboratory, p.Pro59Leu (176C>T; 177G>T). Jurkat cells are an immortalized line of human T lymphocyte cells obtained from the peripheral blood of a boy with T cell leukemia, and they were selected based on their being hematopoietic-derived cell line. However, it is important to note that lymphocyte cells are lymphoid precursor-derived, while the candidate MCTO effector cell types are myeloid precursor-derived cells.
Sophie’s Neighborhood has also partnered with Artisan Bio to generate induced pluripotent cell lines with MCTO pathological mutations introduced. Cell lines containing the mutation that Sophie carries, p.Pro71Arg (CCC>CGC): c.212 C>G, and the mouse model mutation, p.Pro59Leu (176C>T; 177G>T) are both currently under development. The generation of these cell lines will be critical to the development of accurate and informative drug screens, in addition to their use in experiments to better understand MCTO disease mechanisms. While the MCTO effector cell types are currently unknown, the use of iPSCs to generate the possible candidate MCTO effector cell type is advantageous, especially considering that some of the possible candidates do not have well-published immortalized human cell lines available - as is the case for human osteoclasts. However, it is important to point out that, fortunately, several protocols exist for differentiation of iPSCs to the numerous candidate MCTO effector cell types that may be required for future studies, including osteoblasts, osteoclasts, chondrocytes, monocytes, and macrophages.
While the candidate MCTO effector cell types are not currently known for the skeletal pathological phenotypes, the renal phenotype is suspected to be associated specifically with podocytes in the kidney and not be the result of a more systemic effect. As stated previously, the MafB podocyte-specific knockout mice develop a series of symptoms seen in the MCTO mutant mouse, and that phenocopy MCTO human patients, including proteinuria, FSGS lesions, renal interstitial damage, and podocyte foot process effacement (Usui, 2000, Tsunakawa, 2019). Further support of this conclusion is that the MCTO patients who experience kidney failure and received a kidney transplant do not experience a recurrence of renal pathological symptoms after treatment (Matthew Sampson, personal communication).
Podocyte disease modeling is well published, with protocols for 2D and 3D cell culture. The benefit of 3D culture modeling is of course to be able to assess podocytes within kidney organoids in a more accurate representation of how the cells grow and interact within the glomeruli structure. Research has shown that glomeruli from iPSC-derived kidney organoids exhibit improved podocyte-specific gene expression, maintenance of polarised protein localisation, and better glomerular basement membrane matrisome when compared to 2D podocyte cultures. Additionally, organoid-derived glomeruli may be more amenable to toxicity screening, as they retain marker expression in culture for 96 hours (Hale, 2018). However, 2D cell culture systems are typically less expensive, simpler to grow, and often allow for easier cell observation and measurement. 2D podocyte cell culture may be more appropriate for high-throughput drug screening for the drug repurposing track outlined below in the roadmap if Sophie’s Neighborhood decides to pursue additional treatments for the renal pathological phenotypes associated with MCTO.
Lower organism disease models: Zebrafish and Flies (Drosophila melanogaster)
Often Drosophila melanogaster (fruit flies) can be used to expedite drug development by allowing the conduct of large unbiased screens in these animals that have high gene conservation with humans, fast propagation times, and relatively low research costs. However, Perlara does not believe that Drosophila models offer a good opportunity for drug development for MCTO.
The gene traffic jam (tj) encodes the only large Maf transcription factor (MafA, MafB, c-Maf, and NRL) in flies. In tj mutant gonads, somatic cells fail to intermingle and properly envelop germline cells, causing an early block in germ cell differentiation, establishing tj as a critical modulator of the adhesive properties of somatic cells (Li, 2003). In addition, tj is required in pre-adult escort cell (EC) lineage for germline differentiation control (Li, 2019). While the tissues known require Maf function are different in flies vs. humans, this is not necessarily a key roadblock. Instead, it is the lack of conservation of individual Maf homologs (i.e., MafA vs. MafB) that precludes the use of flies for a successful MafB modifier screen and thus investing in Maf Drosophila avatars is not recommended.
In contrast, while Perlara is not recommending zebrafish as a priority research track at this time, Perlara does believe that an unbiased modifier screen of zebrafish MafBB mutants could yield useful information and the potential benefits and pitfalls of such experiments are discussed below.
Zebrafish are excellent disease models for both bone development/disease and kidney development/disease. Zebrafish have large clutch size, are optically transparent enabling real-time observation of many developmental events, and have quick development with all major organs having developed within the first 5 days.
Specifically, osteoclast cell development and skeletal physiology are similar between zebrafish and mammals, due to the transparency of zebrafish embryos, bone development can be easily observed by real-time live imaging (Busse, 2020), and several groups have developed chemical screening platforms for zebrafish bone modifiers (Bergen, 2019, Lleras-Forero, 2020). In the renal system, the zebrafish podocyte has already been well-established as a model for some forms of FSGS and podocyte repair/regeneration (Huang, 2013; Hansen, 2020). Most intriguing, Jens Westhoff and colleagues have established an automated high-content screen to score for phenotypic renal alterations such as glomerular and tubular malformations in order to assess renal toxicity of drugs (Westoff, 2020).
Zebrafish have two mafB paralogs, but mafbb is more preferentially expressed in myeloid lineages and macrophages vs. mafba (Han, 2021). Based on these expression patterns, 2 mafbb knockout (KO) mutant zebrafish mutant lines were made in the laboratory of Lili Jing (Shanghai Jiao Tong University, China) by creating 4 or 11bp deletions which led to an early frameshift mutation. The mafba homozygous KO was viable but had increased mortality (40% at 24 hours post-fertilization), gross but variable skeletal defects (jaw, caudal fin, spine), upregulation of osteoclast development and maturation, defects in bone and cartilage formation, decreased bone volume fraction consistent with osteoporosis, expanded definitive macrophage differentiation, inhibited primitive macrophage development, selective expansion of definitive myelopoiesis including macrophages and neutrophils, and a small reduction of lymphoid and erythroid differentiation.
The similarities of the zebrafish KO skeletal defects and MCTO humans disease suggest that an unbiased suppressor screen of a zebrafish mafb mutant could yield timely and disease relevant pathways that could identify novel pathways to target pharmacologically. However, experiments would need to be done before conducting a screen. Most importantly, a zebrafish MCTO mutation in mafbb has not been made and a screen should be done on a MCTO mutant mafB not the KO. It would need to be confirmed that the mafbb MCTO mutant has skeletal and/or renal defects similar to MCTO patients. Second, if a skeletal screen is planned, additional mouse experiments should be done to narrow down the skeletal cell of interest (see above) in order to select a zebrafish phenotype on which to base a suppressor screen. If a renal screen is desired, the MCTO and KO mafbb mutants need to be assessed for renal defects.
In summary, a unbiased forward genetic screen of a mafbb MCTO mutant zebrafish could be a viable path for MCTO drug development but a cell-based human podocyte screen is likely a faster path for a renal therapeutic given the absence of a mafbb MCTO mutant zebrafish. A skeletal unbiased forward genetic screen in the zebrafish is gated by the lack of understanding of the cell type driving the MCTO skeletal phenotypes in MCTO patients and mouse models. Once a cell type and MafB-dependent developmental/repair process is defined, the suitability of a zebrafish screen can be re-assessed based on the availability of high quality cell-based models.
THERAPEUTIC TRACK 1: DRUG REPURPOSING
Drug repurposing is defined as identifying a new use for an already approved drug, be it a century old drug or a drug that was just recently approved. Drug repositioning is defined as salvaging an experimental drug that cleared the initial hurdle of early-stage safety studies but failed in late-stage efficacy studies for its intended disease indication.
The Broad Repurposing Hub (commercially available as the SPECS library) is an ideal launchpad for identifying drug repurposing and drug repositioning candidates. Any MCTO disease model selected should be screened against this collection as a high-throughput drug screening program.
Selection of a MCTO Drug Repurposing Screening Platform
Given the lack of any therapeutics that reverse or halt the progression of MCTO in the skeletal system and the nearly universal penetrance of skeletal defects among MCTO patients, initial MCTO drug repurposing efforts will focus on the skeletal system. If at a later date, Sophie’s Neighborhood or other foundations chose to pursue a renal MCTO therapeutic, many attractive systems exist for high-throughput screens in renal cell lines (Reiser, 2010, Lee, 2015, Lee, 2018, Hale, 2018).
While unbiased phenotypic screens can be powerful tools to pull out unexpected disease modifiers, a phenotypic screen is not recommended for MCTO until genetic experiments uncover the MCTO skeletal effector cell type(s). Instead a high-throughput screen can take advantage of the strong theoretical evidence for MAFB perdurance being the key biological alteration underlying MCTO pathology to identify drugs which modulate the transcriptional signature of MAFB overperdurance. Arpeggio Bio has developed the PRO-seq Platform to measure nascent RNA (Figure 5) and is currently expanding the technology to a 384-well plate format, which will enable high-throughput screening of cell lines against drug libraries using nascent transcriptional profiling as the readout. This approach is far superior to a more conventional transcriptional screen which would select 1 or 2 known transcriptional MAFB targets as screen readouts as such a screen would have a high risk of selecting uninformative targets that are not central to MCTO pathology - in contrast, a large-scale readout of the MAFB overexpression transcriptional profile will be assessed.
Figure 5: Arpeggio PRO-seq Platform
Source: Arpeggio, 2021
While the MCTO skeletal effector cell type(s) are unknown, this screen could move forward in the near future by selecting one cell from each the hematopoietic stem cell lineage (macrophages, osteoclasts, monocytes) and one cell from the mesenchymal cell lineage (osteoblasts, chondrocytes, and to a lesser extent osteocytes) with selection being based on both biological plausibility of MCTO involvement and ease of cultivation. While patient-derived MCTO iPSC cells are being derived and could be used, iPSC-based screens can take several weeks (or even months) of differentiation per screen and require considerable cell production capacity and lab operational resources. Therefore, an initial screen using a primary cell line overexpressing either Pro59Leu or Pro71Arg MAFB would be a less expensive and more easily produced alternative to iPSCs. Target validation should be performed in the patient-derived MCTO iPSC cells.
Go-no Go Experiments for PRO-Seq-based MCTO Repurposing Screen
Currently, Jurkat cells overexpressing MAFB are being examined in the Arpeggio platform. To prepare for a repurposing screen, Pro59Leu or Pro71Arg MAFB should be expressed in the selected primary cell lines, either constitutively from a vector or introduced into the endogenous locus. MAFB protein perdurance and similar transcriptional profiles to patient-derived iPSC cells should be confirmed.
THERAPEUTIC TRACK 2: Bone Marrow Transplant
In partnership with the Takahashi lab (University of Tsukuba), Sophie’s Neighborhood is evaluating allogeneic hematopoietic stem cell/bone marrow transplantation (HSCT/BMT) as a potential MCTO therapeutic. An allogeneic bone marrow transplant has been performed in the Pro59Leu MAFB mice; the homozygous MCTO mutant mice will be assessed for rescue of the skeletal and renal defects. Results are expected in mid-2022.
The rationale for BMT in MCTO is based on the success of treating selected forms of osteopetrosis with HSCT; osteopetrosis patients with bone marrow failure, aged <1 year at diagnosis, and mutations intrinsic to the osteoclast can be referred to a specialty center for consideration for HSCT (Wu, 2017; Schultz, 2015). HSCT is the only curative treatment for the infantile malignant form of OP, and some patients with intermediate OP who suffer from debilitating skeletal complications of OP have also experienced dramatically improved symptoms and quality of life post-HSCT (Stepensky, 2019).
Currently, Sophie’s Neighborhood SAB disease experts are divided on how seriously a HSCT/BMT should be considered for the average MCTO patient, including Sophie. While some SAB members believe that the real but known risks of BMT compare favorably to the unknown risks of a novel therapy, other SAB members are concerned that the likelihood BMT reversing or correcting the abnormal bone development in an already symptomatic child are low and argue that the risk:benefit profile may not be in favor of BMT. Most of Sophie’s Neighborhood SAB disease area experts believe that BMT will likely only affect the skeletal aspects of MCTO.
If the BMT mouse results are positive in mid-2022, Sophie’s Neighborhood will need an all-hands-on-deck meeting to wrestle with whether to move forward with a BMT MCTO clinical trial. During such a discussion key questions will include:
How does the timing of the BMT compare with the proposed treatment timing in a MCTO patient? Do the results suggest that abnormal bone development can be corrected or that further deterioration of unaffected joints can be protected?
Based on any additional knowledge of the basic mechanisms of MCTO in summer of 2022, are there any additional histological or other experiments that can be done on the transplanted mouse limbs to further tease apart which cell types and aspects of MCTO are affected by BMT?
What conditioning protocols should be considered for BMT for MCTO? What impact could be anticipated on the kidneys?
Given that there is little expectation of a positive effect of BMT on the renal aspects of MCTO (though this experiment will be done in the mice), what is the impact of a BMT on the feasibility of future renal-targeted MCTO therapeutics?
THERAPEUTIC BACKUP TRACK 1: MAFB ASO, allele-specific or directed to all MAFB transcripts
Given that MCTO is likely caused by overactive MAFB, an antisense oligonucleotide-based therapy (ASO) is a compelling approach in which an ASO could reduce the abundance of either all MAFB transcripts or MAFB transcripts specifically carrying Sophie’s mutation in a local area of the body.
An allele-specific ASO is attractive because the wild-type MAFB protein, which should be turned over in a physiologically appropriate manner, would be preserved, while the mutated MAFB protein would be dramatically reduced. Haploinsufficiency in MAFB is known to cause Duane Retraction syndrome, an eye movement disorder sometimes associated with FSGS (Kaimori, 2018, Sato, 2018), and given the diversity of systems MAFB acts in, dramatic reduction of MAFB levels would be expected to lead to other serious adverse effects. Therefore, preserving the wild-type MAFB transcripts is attractive, but to date, even highly selective allele-specific ASOs in clinical development, still reduce the wild-type transcript to some level (Timothy Yu/Boston Children’s Hospital, personal communications). Therefore even in the case of an allele-specific ASO, experiments would need to be conducted, likely in both cells and mice, to determine the acceptable ranges of MAFB reduction, both in the case of wild-type MAFB and animals and cells that are MAFB MCTO carriers. It is also important to keep in mind that development of an allele-specific ASO compared to conventional ASO development is expected to be a significantly more lengthy and expensive process given the need for multiple rounds of screening for allele-specificity. In addition, anecdotal evidence from labs which are developing allele-specific ASOs has shown that allele-specific ASO often require novel, non-clinically validated nucleotide chemistries, which have less established safety and delivery profiles (Timothy Yu, personal communication). Finally, an allele-specific ASO therapy would likely only be able to be used to treat Sophie, as no other MCTO patients are yet known with the same MCTO mutation.
Given that overactivity of MAFB is the likely cause of MCTO, an allele-specific ASO may not be necessary, as knock-down of either mutant or wild-type MAFB activity would be expected to improve symptoms. This approach would be more affordable, timely, and would also allow the development of a therapeutic that could improve the lives of all MCTO patients.
A major issue of an MCTO ASO therapeutic would be delivery, but depending on the targeted skeletal cell types, it could be feasible to deliver the ASO as an intra-articular injection in the affected joints, similar to a steroid injection. Recently, locked nucleic acid (LNA)-based ASOs have been tested in vitro and in vivo for their potential in osteoarthritis (OA) and shown some promising results in joint-protective effects in OA animal models (Nakamura, 2020), suggesting the potential for this approach in MCTO. In addition, Causeway Therapeutics is developing a miRNA therapy OsteoMiR for OA that they plan to deliver by intra-articular injection. Finally, it is worth noting that ASOs targeted at Notch2 have been developed for treatment of the osteolytic disease Hajdu-Cheney syndrome, and subcutaneous administration of the ASO down-regulated Notch2 ameliorates the cancellous osteopenia in mice (Canalis, 2020). These results in Hadju-Cheney are an important proof-of-concept that ASOs can be taken up by key skeletal cell types in the mouse, however, systemic administration of an ASO in humans is not advisable, so this represents early stage work on an ASO therapy for Hadju-Cheney.
Finally, it is important to note that at least one MAFB expert, Roland Stein (Vanderbilt), has expressed skepticism about the potential of an ASO approach for MCTO. He is concerned that at least in the case of MafA that the transcripts are not the limiting factor and that given the expected very long persistence of mutated MAFB protein in MCTO, it may be difficult to knock-down MAFB activity enough to see an effect in MCTO. For that reason, a MAFB targeted protein degrader is also proposed (see next section).
THERAPEUTIC BACKUP TRACK 2: Targeted Protein Degrader
Given the concerns about the potential for extreme purdurance of MAFB protein in MCTO, targeted protein degradation is an alluring but somewhat risky potential alternative to an ASO approach. In TPD, a proteolysis-targeting chimera (PROTAC), uses the cell’s ubiquitin proteasome system to selectively degrade the protein of interest. One end of the small molecule binds to an E3 ubiquitin ligase, while the other end attaches to the protein target of interest, triggering the transfer of a ubiquitin chain to the target protein.
TPD is an attractive modality for MCTO because it is the MAFB protein that is modulating MCTO pathogenesis and because MAFB is already known to be ubiquitinated. However, while there are an ever growing number of TPD clinical programs, TPD is still in its infancy as an established drug development pathway, and lead development workflows, toxicology and off-target parameters are still being worked out (Garber, 2021). Therefore, TPD is suggested as a backup therapeutic track to consider, especially as the TPD knowledge is expected to grow exponentially in the next several years.
THERAPEUTIC SHORT-CUT 1: 3’SL MCTO Therapeutic Candidate
Identification of 3’SL as Potent MAFB Suppressor
As of November 2021, the Arpeggio database held nascent RNA sequence data from >1,000 treatments, 3,500 samples, and 50 cell types. As a proof-of-concept, the Arpeggio database was queried for compounds that inhibit the transcriptional activity of MAFB and 3’-Sialyllactose (3’SL) was identified as a potential and potent inhibitor of MAFB-dependent transcription in M1 macrophages (Figure 6).
Figure 6: Arpeggio PRO-seq Database Identifies 3’-Sialyllactose as Potent MAFB Inhibitor in M1 Macrophages

Source: Arpeggio, unpublished data, 2021
3’SL is an attractive candidate therapeutic for MCTO because of it’s high biological plausibility as a MAFB inhibitor in the tissues of interest, strong, established safety profile at least the dose levels examined for human use as a food additive/supplement, expected commercial availability in the near future, and the ease of systemic delivery.
3’SL’s Role As An Immune Modulator and Commercial Development as a Novel Food
3’SL is a sialylated human milk oligosaccharide (HMO) which is abundant in human milk, and which can be obtained by fermentation and isolated as a purified ingredient in the sodium salt form (EFSA, 2020). 3′SL is known to regulate immune homeostasis in a variety of tissues (Zenhom, 2011; Kang, 2020) and has been suggested as a potential modulator of metabolic/cardiovascular health (Harris, 2020). Most intriguingly, 3’SL has been shown to suppress interleukin-1ꞵ-induced cartilage matrix degradation in chondrocytic cells, by increasing levels of enzymatic antioxidants, reverse the levels of enzymatic antioxidants, inhibit the apoptotic process, and reverse cartilage destruction by decreasing the release of matrix metalloproteinases such as MMP1, MMP3, and MMP13 (Baek, 2021). The correlation of 3’SL effects on cartilage matrix degradation in chondrocytes and the inhibition of apoptosis raises the possibility that some of these effects may be mediated by MAFB and that 3’SL should be tested as a MCTO candidate therapeutic.
Notably, the global nutrition company DSM is developing 3’SL as a human food additive and supplement for the US and EU markets and obtained a favorable opinion from the European Commission’s EFSA Panel on Nutrition, Novel Foods, and Food Allergens for the use of 3’SL as a novel food in the EU in 2020. DSM has developed a Good Manufacturing Practice (GMP) and Hazard Analysis Critical Control Points (HACCP) compliant manufacturing process for 3’SL and was originally hoping to launch 3’SL in the US and EU markets in 2020, though launch may have been delayed due to the COVID pandemic. While DSM has proposed a maximum daily intake of 3”SL of 0.5g/day, which is equivalent of a maximum daily intake ranging from 7 to 22mg/kg bw, a 14-day repeated dose oral toxicity study in rats concluded that a high dose of 5,000mg/kg bw/day was considered a the NO Observed Adverse Effect Level (NOAEL). A bacterial reverse mutation test and an in vitro mammalian cell micronucleus test of 3’SL revealed no concerns regarding genotoxicity of 3’SL.
Experiments to Establish 3’SL as a MCTO therapeutic candidate
If the basic science of 3’SL as a MCTO suppressor is verified, 3’SL could be developed as a MCTO therapeutic within a relatively short timeframe. First, 3’SL ability to inhibit MAFB-directed transcription should be confirmed in the best currently available cell model, assessing both MCTO mutant cell lines as well as a line overexpressing MAFB. Next, MAFB inhibitory dose ranges should be established in a MCTO chondrocyte line, given the proposed role of the chondrocytes in MCTO pathology and the established role of 3’SL in regulating key inflammatory pathways in these tissues. Finally, several doses of 3’SL should be tested in the Pro59Leu MCTO mouse model for modification of the skeletal and renal MCTO defects.
Figure 7: Schema for 3’SL Development as a Candidate MCTO Therapeutic

THERAPEUTIC SHORT-CUT 2: Senolytics/Senomorphics
Does Premature Senescence Contribute to MCTO Pathogenesis?
Recent observations that the mutations causing persistence of the highly related transcription factor MafA induces premature cellular senescence through the activation of senescence-associated secretory proteins (SASPs) (Walker, 2021), has raised the question of whether MCTO pathogenesis could be senescence related and, thereby treated with senolytics. Cellular senescence, a durable, cell cycle arrest response that regulates cell fate specification/patterning and function in both developing and mature tissues, is induced in a variety of disease states (Gorgoulis, 2019). In support of a possible role of senescence in MCTO pathogenesis, overexpression of MAFB in Jurkat cells cells causes slightly decreased cell count and the effect is greatly magnified when either the Pro71Arg or Pro59Leu mutations are introduced into the overexpressed MAFB (Tanya Warnecke, unpublished results, 2021).
Go-no Go Experiment for Senolytic/Senomorphic for MCTO
The limbs from Pro59Leu MCTO mice can be stained with senescence-associated β-galactosidase (SA-β-gal) and compared to wild-type limbs at various developmental ages to establish whether premature senescence is activated in MCTO. This straightforward experiment would be a go/no-go assay for further investigation into senolytic (agents that selectively clear senescent cells) / senomorphic (agents that delay or prevent senescence) therapy for MCTO. If positive, this initial result would open up the development of a senolytic/senomorphic therapy for MCTO, a promising and burgeoning field in drug development for diseases such as atherosclerosis, osteoarthritis, osteoporosis, cancer, diabetes, neurodegenerative diseases, and cardiovascular/metabolic diseases (Lagoumtzi, 2021; Ellison-Hughes, 2020; Dolgin; 2020), many of which are already in clinical development. In addition, localized senescence in mouse limbs could point towards a specific cell type responsible for the MCTO skeletal phenotype, providing early direction towards a specific skeletal cell type before more definitive genetic results are available.
THERAPEUTIC BACKUP SHORT-CUT 3: Mining Natural MCTO Human Variation
Most MCTO cases arise spontaneously with neither parent carrying the mutation, although approximately one-third of cases are familial with parents also experiencing either renal or skeletal disease. In addition, a scan of >900,000 patients from the Regeneron and UK Biobank databases found no mutations in MAFB loci associated with MCTO (Sophie’s Neighborhood, unpublished data), suggesting that MCTO-causing mutations are ultra-rare and typically associated with symptomatic disease. However, there is a single report of a family in which a 17-year-old boy with severe carpal-tarsal osteolysis (symptoms originally presenting at age 4), Arnold-Chiari malformation type I, but no renal dysfunction was diagnosed with MCTO after discovery of a MAFB Ser56Phe mutation (Dworschak, 2013). Intriguingly, his mother, 20-year-old sister, and maternal grandmother all carry the Ser56Phe mutation but do not have any symptoms of MCTO, including lack of more subtle abnormalities by wrist x-ray, renal, and urine analysis of the mother. This family is the only report of incomplete penetrance of the MCTO skeletal phenotype, with the exception of a second case report in which an affected child had a father without skeletal abnormalities (Narhi, 2021), but this case was explained by mosaicism, not incomplete penetrance.
The Dworschak incomplete penetrance case offers a unique opportunity to identify naturally occurring and physiologically safe suppressors of the MCTO mutation. The severe skeletal abnormalities in the affected male and the complete absence of skeletal abnormalities in the females, suggests that the suppressor could be X-linked but the very small sample size could make this gender disparity a spurious finding. MCTO cases are evenly divided among males and females (18:15 male: female; Table 1), suggesting the gender disparity is not due to mutant MAFB. Finally, it is possible that the incomplete penetrance of the Ser56Phe mutation is due to environmental modification, but the severity of the phenotype in the affected male, the likely similar environment of the unaffected sister, and the absence of any other reported incomplete penetrance cases argue for a genetic suppressor.
If the family reported in the Dworschak paper consented to providing fibroblasts from the affected son and an unaffected related carrier (ideally mother or sister) for study, these fibroblasts could be differentiated into a skeletal cell type(s) of interest (ideally the cell types chose for further study in Therapeutic Track 1, with a cell type from the macrophage/monocyte lineage and and one from the mesenchymal osteochondral- progenitor lineage) and transcriptionally profiled and compared to either Sophie’s differentiated cells, cells over-expressing MAFB, or cells expressing the Pro59Leu MAFB. This analysis could identify signalling pathways in which alterations may suppress the skeletal abnormalities from MCTO causing MAFB mutations which are well tolerated in the human body. Pathways which are altered in the unaffected sister/mother but not the affected son would represent candidate pathways for modulation by a drug therapeutic, which could have a more favorable safety profile versus drug candidates identified by other methods. Given the very small number of samples to compare, genomic analysis was not considered feasible for identifying the MCTO suppressor, given the expectations of a high signal-to-noise ratio in the analysis of this very small sample set.
Importantly, the effect of the Ser56Phe mutation on MAFB overexpression/persistence in heterologous cells should be assessed prior to contacting the Dworschak patient family to confirm that that the Ser56Phe mutation leads to MAFB overexpression/ persistence, given that this mutation has not be reported in other MCTO cases. Then prior to transcriptional analysis, and possibly cell derivation, if MAFB levels can be assessed in fibroblasts, it would be important to assess whether MAFB protein levels or turnover are affected in the Dworschak patient samples. This experiment would determine both whether the suppression is occurring upstream or downstream of MAFB overexpression, and confirm that the Ser56Phe mutation is causing MAFB overexpression/persistence in the affected patients’ cells.
CLINICAL TRIAL READINESS
Natural History Study
Although a systematic review of the MCTO literature published in October 2021 yielded valuable insights into disease progression, such an approach is strongly impacted by reporting bias (i.e., absent symptoms are not reported and therefore the prevalence of less common symptoms may be over-reported, although the converse may also occur). Therefore, a carefully crafted longitudinal natural history study is required in order to create comparator groups to aid in clinical trial design and render randomized placebo-controlled trial designs unnecessary.
Such a natural history study is expected to initiate at the University of Colorado under the direction of Dr. Nina Ma in the near future. This study will create an invaluable dataset, showing the inter-patient and intra-patient variabilities in disease presentation in order to guide trial design and discussions with regulators. Data over 2-3 years from 7-10 MCTO patients could be a game changer.
The registry will be comprised of retrospective/prospective chart review, including local care, birth details, medical/ surgical history, genetic results,radiographic information including standardized, central image assessment (number of ossification centers and number and types of bones impacted). Longitudinal centralized image assessment will allow the study to address novel questions in MCTO, including delineation of defects due to abnormal bone development versus osteolysis in affected joints and in particular looking at both the CT and large joints (knees and elbows) to assess whether development is impaired and whether the joints develop arthritis,inflammation-directed osteolysis, or primary osteolysis.
Patient-reported outcome measures (PROs) will also be assessed, but a MCTO-customized questionnaire needs to be developed. The questionnaire can be extrapolated from other genetic and musculoskeletal disease such as osteogenesis imperfecta (OI), which has validated the use of the Pediatric Outcomes Data Collection Instrument (PODCI) in pediatric OI patients (Murali, 2020). PODCI has also been validated for the use of assessing pediatric patients receiving hand therapy (Dorich, 2019) and a similar PRO has been assessed for pediatric shoulder and elbow disorders (Heyworth, 2018). Alternatively, the CAPTURE-JIA patient-reported outcome measures (PROM) and patient-reported experience measures (PREM) patient-reported outcomes measures were recently piloted and validated in a 2-center study as an improvement over JIA questionnaires used for clinical research that do not always function well in pediatric populations (Lunt, 2020).
Fortunately, there is no need to reinvent the wheel in terms of data sharing platforms. The AT Children’s Project whole genome sequencing collaboration with the Broad Institute serves as a model for data sharing. The nonprofit foundation RARE-X would make an excellent partner for a pilot project focused on MCTO. Citizen, which was recently acquired by Invitae, is a digital patient registry platform and electronic medical records aggregator that could also be a potential collaborator. AllStripes is another for-profit company that works with patient communities to aggregate and standardize electronic health records as well as offering an out-of-the-box natural history study survey/questionnaire product to help groups like SN build patient-reported outcome measures (PRO’s) that would pass muster at FDA.
MCTO Clinical Trial Design
This roadmap envisions several single-patient observational and safety “pioneer” studies for MCTO, followed by a larger clinical trial in 10-15 MCTO patients. Given the small size of the worldwide MCTO population (~30 patients), the natural history study will establish comparator group data that will render randomized placebo-controlled trial design likely unnecessary.
Once a MCTO therapeutic candidate is identified, potential disease partners should be immediately identified based on the pathway targeted. If the identified MCTO candidate therapeutic is thought to have the potential to modify closely related ultra-rare orphan diseases such as Multicentric osteolysis, nodulosis and arthropathy (MONA), it may be possible to run parallel trials, or even separate arms of the same larger trial, in order to share trial logistics and/or funding mechanisms across diseases. If the pathway targeted by the candidate MCTO therapeutic is expected to have potential in a common disease such as another bone/cartilage disease, or even immune/renal/metabolic disease, partnering activities should be promptly initiated in order to identify funding opportunities and to hopefully spur the development of a program that would generate a larger safety database for the candidate therapeutic in both adult and pediatric populations.
MCTO Clinical Trial Endpoints
MCTO clinical trials will need to probe endpoints that account for the different disease progression rates among patients enrolling at different ages and disease stages and will need to capture both key patient-reported outcome measures to reflect improvement in activities of daily living and clinical measures of anatomical disease progression.
In the kidney, potential MCTO renal endpoints are straightforward, with proteinuria (percent change in urine protein/creatinine ratio) being established as an acceptable primary endpoint in FSGS (current pediatric FSGS trials). Historically, complete remission of proteinuria was the goal, but a ~30-40% reduction of proteinuria with preservation of estimated glomerular filtration rate (eGFR) is now an accepted endpoint in FSGS (DeVriese, 2021; Troost, 2020). Change in eGFR should also be assessed as a separate secondary endpoint. In addition, consideration should be made if patients with severe renal dysfunction (eGFR<30ml/min/1.73m2) should be excluded from the renal analysis, as these patients may be expected to progress into renal failure regardless of MCTO-specific intervention.
Skeletal endpoints which should be considered for MCTO clinical trials include: time to loss of ambulation, joint mobility, pain measures, number of ossification centers in the CT and possibly knee and elbow joints, number of carpal-tarsal bones, bone density (not expected to be affected in all patients), volumetric measurements of specific CT bones and/or lesions, and the patient-reported outcomes score developed for the natural history study. Measuring bone lesions by MRI would be superior to CT because of the reduction in radiation load of the patients and MRI can also detect bone marrow edema, which is an antecedent to osteolysis.
The primary endpoint should be able to capture disease events which are expected in all MCTO patients and which can be robustly measured in a reasonable timeframe. Therefore, the primary endpoint should focus on measures of the carpal-tarsal, and possibly knee and elbow, lesions. As patients will be enrolled at different stages of development and disease progression and the disease progression and therapeutic modification expected in 2 year olds and teenagers are quite different, the primary endpoint will have to account for different expected outcomes based on age. As there is no established method for scoring these lesions, early conversations with the FDA will be necessary to ensure alignment on the primary endpoint. Other skeletal diseases such as fibrous dysplasia, articular cartilage lesions/osteoarthritis, and multiple myeloma have developed methods to measure other skeletal lesions (Table 5) that may be used to guide MCTO-specific measurements.
Table 5: Clinical Trial Primary Endpoints in Selected Osteolytic or Related Diseases
Disease Trial Primary Endpoint

No trials were registered at clinicaltrials.gov for the related diseases of Hajdu-Cheney; Gorham-Stout (no skeletal trials); Melorheostosis
Pre-IND Meeting
The natural history study and initial preclinical proof-of-concept experiments to guide therapeutic candidate selection will build a foundation for a pre-Investigational New Drug (IND) meeting request for the treatment of MCTO sometime in the first half of 2023. Feedback from the pre-IND meeting will provide increased clarity on what kind of clinical study will best serve MCTO patients in the development of therapies.
BIOTECH READINESS
Biotech readiness starts with identifying biopharma partners or venture philanthropy partners as potential sponsors of specific therapeutic tracks. Initially, funds will have to be raised (or donations granted) for communal resources like disease models and datasets. Once those capacity-building and platform-enabling projects are completed and limited preclinical proof-of-concept experiments point towards a specific therapeutic track or tracks for MCTO, Sophie’s Neighborhood will be in a strong position to negotiate with biotech startups in exchange for access to data, assets, and IP generated with foundation support.
Potential overlaps between MCTO and larger disease populations
Given the promiscuity of MAFB in regulating a diverse set of target genes in a wide variety of tissues, the sky's the limit on the number of rare and common diseases that could be treated with a therapeutic initially discovered in MCTO research. Roles for MAFB have been suggested in diverse diseases such as atherosclerosis (Hamada, 2014), obesity-related diseases (Pettersson, 2015), oncology (Chen, 2020), diabetes (Conrad, 2016; Cyphert, 2019), in addition to the established role in the renal and skeletal systems affected in MCTO.
Developmental milestones which will uncover potential partnering opportunities
Partnering with other foundations and biotech companies to develop a MCTO therapeutic for broader indications beyond MCTO will not only unlock additional funding opportunities, but will also allow the assessment of safety for MCTO candidate therapeutics in much larger patient populations. Therefore, potential of any candidate MCTO therapeutic in non-MCTO indications should be assessed through conversation with disease area experts and potential biotech partners early and often. Once a go-decision has been made for further characterization of a candidate MCTO therapeutic after initial preclinical proof-of-concept experiments have assembled a small package of compelling data (see example for 3’SL), partnering and information gathering conversations should be initiated.
FUNDING MODEL
Sophie’s Neighborhood will provide the seed money for Year One activities described above. Data generated from these activities will then serve as a basis for the fundraising necessary for subsequent drug development.
OPERATING STRUCTURE
Sophie’s Neighborhood has established a strong Scientific Advisory Board to guide initial decisions for MCTO therapeutic development. Once specific therapeutic candidates are identified, and in parallel the consideration of impacts on larger disease populations, Sophie’s Neighborhood may find it advantageous or strategically aligned to create a separate company or entity, operated jointly with Perlara, whose purpose is to raise and manage an umbrella fund or series of programmatic funds that makes milestone-based early-stage startup investments across the MCTO therapeutic landscape.
In this vein, Sophie’s Neighborhood may want to examine funding and operating structures from other rare diseases in which the patient organizations receive economic value for the data that can then be reinvested in the mission. Examples of foundation-run venture funds include:
Juvenile Diabetes Research Foundation T1D Fund - $125M raised from donors who get tax benefits + co-investment rights
CureDuchenne Ventures – funded from their share of proceeds from Prosensa’s sale to Biogen after funding exon-skippers
Myeloma Investment Fund – $50M run by Multiple Myeloma Research Foundation with donor money
METHODOLOGY
This MCTO Cure Roadmap was prepared between October 2021 and January 2022. Perlara PBC received documents from Sophie’s Neighborhood outlining current and planned research activities. After reviewing the MCTO/MAFB primary literature, Perlara conducted interviews with Sophie’s Neighborhood disease area experts and SAB members, Nina Ma (Children’s Hospital Colorado, Endocrinology), Matthew Sampson (Boston Children’s Hospital, Nephrology), Craig Forester (Children’s Hospital Colorado, Hematology), and Michael Levine (University of Pennsylvania, Endocrinology), as well as key scientific collaborator Satoru Takahashi (University of Tsukuba, mouse geneticist and Maf expert). Exploratory calls with potential scientific collaborators Arpeggio Bio, Talus Bio, and Vineet Gupta and Jochen Reiser (both Rush University, Nephrology) were conducted with Perlara and Sophie’s Neighborhood. Informational interviews were conducted with outside scientific experts Syndia Lazarus (Queensland Health, Endocrinology, MCTO expert), and Roland Stein and Jeeyeon Cha (MafA/B experts, Vanderbilt University). Lauren Rosenberg, Sophie’s mother and chair of Sophie’s Neighborhood, was asked to prioritize therapeutic goals for Sophie’s MCTO treatment. Following these discussions and research, the MCTO Cure roadmap was drafted. Reviewers X, X, and X provided feedback on the draft and the MCTO Cure Roadmap (?January 2022 version?) was finalized on X.
ROADMAP UPDATES
This Roadmap is considered version 1 (v1) and it will require annual comprehensive reviews to incorporate progress reports and course corrections.