Repurposing is a gateway drug
A drug repurposing project for the ultra-rare mitochondrial disease MEPAN has yielded a clinical candidate and serendipitously illuminated a pathway to designer pharmacological chaperones using GenAI.


In collaboration with



Disclaimer
The results of the MEPAN drug repurposing project that we are sharing in the spirit of open science below are novel preclinical research findings and therefore they do not constitute the practice of medicine. Please consult a physician or clinical care team if considering off-label use of any approved drug or compassionate use of any experimental drug. The same caution applies to nutraceuticals, supplements and “generally recognized as safe” compounds.
According to prevailing TradBio tastes, drug repurposing is the unsexiest therapeutic modality and rare diseases are the least desirable — let’s face it, money-making — patient populations. Now try getting Pharma jazzed about drug repurposing for rare diseases. Good luck with that. We all know why rare diseases don’t attract Pharma’s gaze: they’re just numerically too small in an era when global blockbusters for chronic diseases of aging like Ozempic, and its entourage of fellow GLP-1 agonists, have made a comeback. Statins 2.0, you could say.
The double irony is that Pharma doesn’t seem to be aware of its own history. Both statins and GLP-1 agonists were originally developed for other indications, and in the case of statins a smaller indication, before expanding to their current mega blockbuster reach. Statins were originally approved in 1987 for patients living with refractory high LDL levels caused by familial hypercholesterolemia. In 1998, the first statin was approved for primary prevention of coronary heart disease. In 2024, 1 in 4 people in America takes a statin.
GLP-1 agonists were originally approved in 2005 for treatment-resistant type 2 diabetes. Nine years later, the label was expanded for the management of obesity in 2014. Today, it is estimated that 1 in 8 Americans is on a GLP-1 agonist. The industry and media are heralding GLP-1 agonists as the first longevity drugs.
But it’s not just economics that retard the potential of drug repurposing. The principal non-economic reason why drug repurposing is overlooked is the impression that the best outcome patients can hope for is a symptomatic treatment that compensates for disease sequelae without snuffing out the fire at its source. In some cases, that’s true. Yet the same can be lamented of recently approved drugs, especially in cancer and Alzheimer’s, that are disease modifying but not disease ending.
In the positive telling, drug repurposing is about getting a runner on base. First base will do. The expectation is not a walk-off grand slam. Those high hopes are usually exclusively reserved for targeted therapies that fix the genetic lesion in the DNA or restore function to the lesioned protein.
The unexpected results of the MEPAN drug repurposing project demonstrate that drug repurposing is underestimated at our collective peril. Like in almost every other drug repurposing project we’ve run or helped manage — 26+ different rare disease programs over the last 5 years — screens powered by patient avatars or patient fibroblast led to the discovery of at least one compelling clinical candidate, and usually several actionable contenders that share a common mechanism of action.
Just our yeast-powered screens alone have a 90% conversion rate from screen to pioneer, parent-led n-of-1 studies. So any argument that drug repurposing is a desperate fishing expedition can be handily dismantled by the evidence amassed by Perlara alone, let alone countless other drug repurposing success stories made possible by others. Yet one is within their rights to steelman the other side and pose the following pointed questions:
When treating older children where the disease is entrenched and some damage is irreversible, can we expect a repurposed drug — either as a monotherapy or multiple compounds stacked in combination — to provide meaningful benefit?
Can a repurposed drug be expected to target the root cause of a disease it wasn’t originally developed for?
Will a repurposed drug just activate compensatory bypass mechanisms and therefore quickly hit an efficacy ceiling?
What are the chances that polypharmacological lightning strikes and drug repurposing becomes a gateway to novel drug discovery?
We all agree that if a treatment is started early enough, it can snuff out pathophysiology in the womb and prevent the disease from even manifesting. Take the example of a fetus with cystic fibrosis (CF) that was washed in Vertex’s Trikafta in utero and was born without classic CF symptoms. Several similar case studies have been reported.
Vertex spent years and billions of dollars developing their fleet of targeted CF small molecule designer drugs, starting with Kalydeco (ivacaftor), the inaugural pharmacological chaperone to be approved for CF after the idea was first proposed in the early 2000s. Shown below is a crystal structure of CFTR bound to second-generation and third-generation CFTF correctors, the culmination of thousands of optimization cycles.

Every protein implicated in a rare disease should be able to receive the CFTR treatment. But it can’t take $10,000,000,000+ and over a decade per program. Times have changed since the idea for a pharmacological chaperone was born two decades ago and when GPUs were just powering graphics cards for video games.
Fast-evolving and rapidly improving GenAI will accelerate the development of designer small molecule chaperones, small molecule correctors, small molecule potentiators — small molecule “you-name-its” depending on how it salves the broken protein. What we found by equal part luck and persistance is that a drug repurposing hit could be the seed for GenAI-enabled designer drug discovery.
The pantheon of CF designer drugs proves that in theory we can scale the development of designer pharmacological chaperones and more expansively designer small molecules that bind to a mutant protein and counteract or reverse the effects of a pathogenic mutation on protein function. Self-proclaimed AI-first drug discovery companies like In Silico Medicines and Excistencia have taken AI-designed drug candidates into the clinic but not yet across the finish line.
Chemical space is certainly large enough that a molecule exists for every mutant protein. Vertex resorted to brute force unbiased phenotypic screening, which don’t get me wrong is the tried and true methodology. Drug repurposing can take advantage of unbiased phenotypic screening too, but instead of a library of novel lead-like compounds, the starting point is a library of repurposable compounds. Using yeast avatars of MEPAN, we did just that. Turns out the seed phrase for MEPAN is echinocandin.
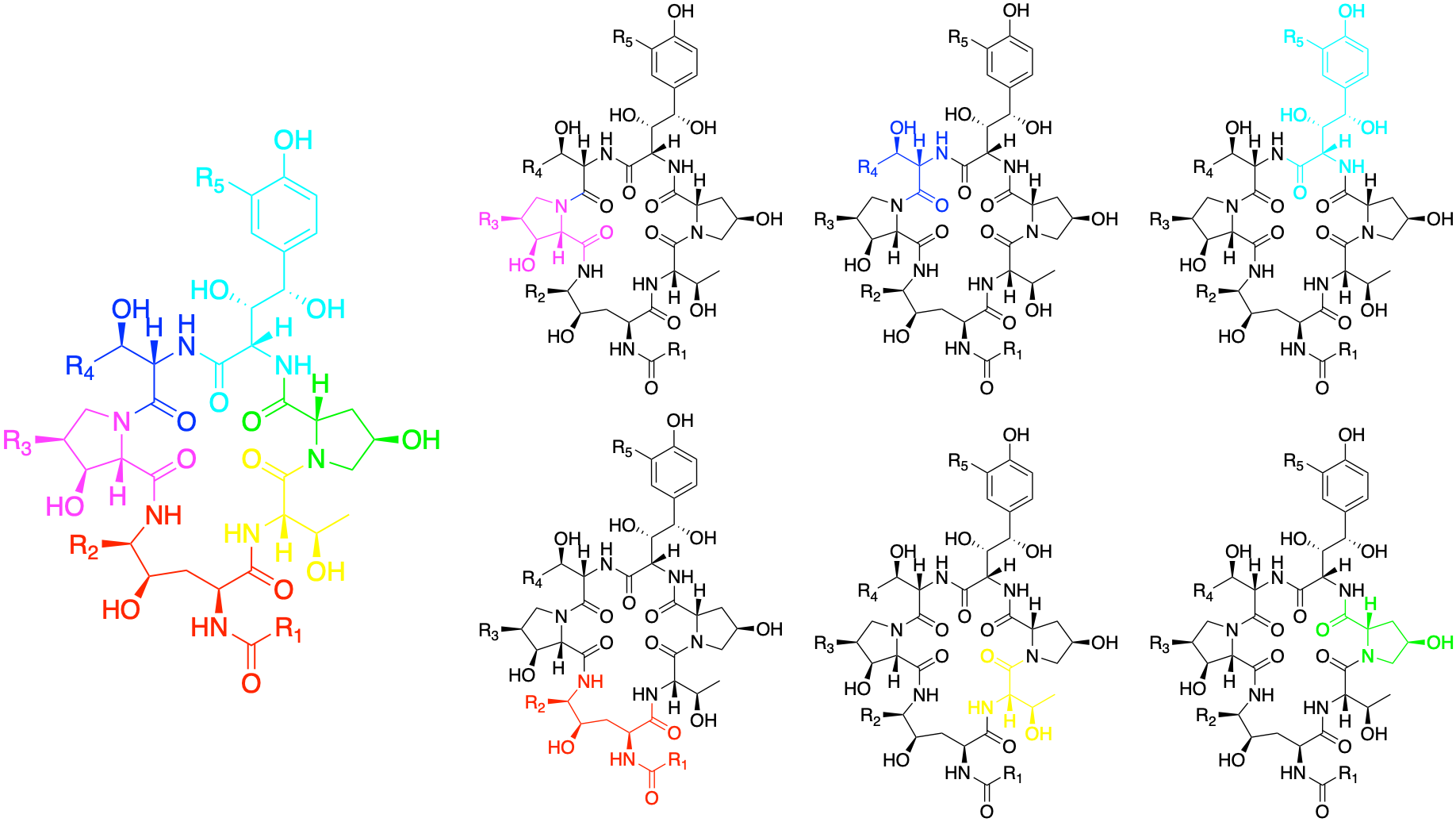
Echinocandins are natural products that humanity turned into antifungals. What jumps off the page is that they are cyclic peptides: larger than conventional small molecules, but still dwarfed by biologics like antibodies. As shown in the figure above, an echinocandin molecule is comprised of six amino acids (each rendered in a different color). The R groups can be derivatized as well.
Peptides are miniature proteins. They can fold. They adopt conformational states. The on-target for echinocandins is a glycosyltransferase that is essential for the assembly of the yeast cell wall. As previously described here, the top hits from the yeast-powered MEPAN drug repurposing screen are caspofungin, micafungin and anidulafungin. We were at first struck by the fact that antifungals of wildtype yeast were acting as rescuers of MECR-deficient yeast. Wouldn’t the antifungal effect of echinocandins swamp out any other secondary pharmacology or providential off-targets?
We in fact see that with caspofungin, the first-generation echinocandin and also the dirtiest with the simplified acyl tail, has an upside down U-shaped dose response. The antifungal effect does eventually swamp out the MECR rescue effect, but interestingly in the second-gen micafungin and third-gen anidulafungin, that high dose tox is gone.
Critically, only MECR missense mutants that still create a folded protein are rescued by echinocandins. There was no rescue observed in the MECR knockout, demonstrating that at least a quasi-stable full-length MECR protein is required for the echinocandin rescue effect. The killer experiment will be to measure the binding of echinocandins to different variants of the MECR protein. For now, the most parsimonious explanation is that the echinocandins bind to the MECR protein and are acting as unoptimized MECR pharmacological chaperones.
How could that even be possible? Let’s take a close look at the echinocandin structures and in particular the modifications between them as the chemical series was optimized for antifungal activity and superior pharmacokinetic properties over time. Rezafungin differs from anidulafungin by the addition of a single choline group at the R2 position.

The ultimate test is to apply the echinocandins in a setting where the on-target antifungal target is off the table, i.e., the yeast target of echinocandins does not exist in human cells. So in the absence of a fungal infection, enchinocandins should be inert in the human body. Therefore we tested the echinocandin series on MEPAN patient fibroblasts and focused on mitochondrial function as a readout using Seahorse assays.


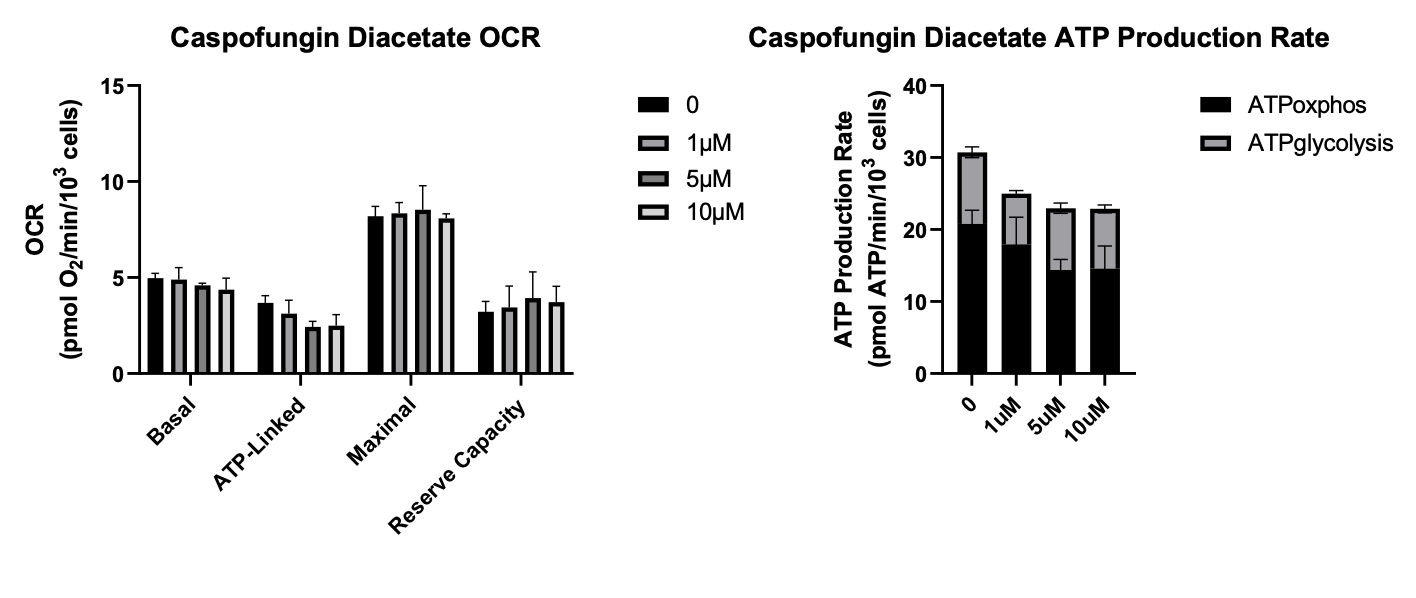

Based on OCR (oxygen consumption rate) and ATP production rate, anidulafungin rose to the top of the pile, followed by micafungin in not-too-distant second. With micafungin, the uptick in ATP production rate at the lower doses disappears at the highest dose. Caspofungin was toxic from the get-go, reducing ATP consumption rate at all three doses tested, and it had no effect on OCR.
Rezafungin to our mild surprise did not have any effect on OCR but did seem to induce a bump in ATP production rate. The same rank order of rescue we observe in the MECR yeast avatars is recapitulated in MEPAN patient fibroblasts. Rezafungin and anidulafungin differ by a single substitution that presumably makes rezafungin an even better antifungal but a slightly worse MECR chaperone. Even with just four members of a chemical series, a structure-activity relationship between the echinocandins and pharamcological rescue of MECR is starting to emerge.
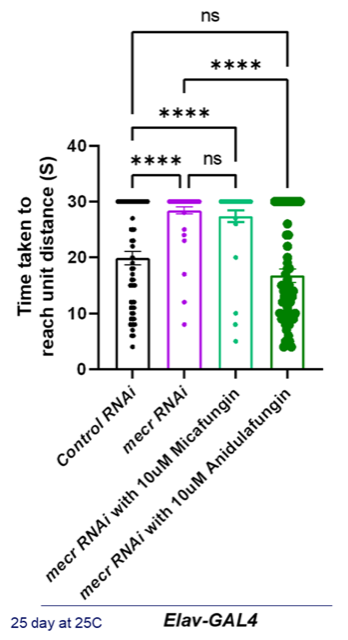
We have evidence from yeast. We have evidence from fibroblasts. But no data from an organism establishing rescue of a non-cellular phenotype. In collaboration with Prof. Hugo Bellen’s lab, micafungin and anidulafungin were evaluated on a fly model of MEPAN that they generated. As shown in the figure below, anidulafungin rescues a wall climbing behavioral phenotype in the fly, but micafungin does not.
Altogether, we think the data package for a single-patient IND with anidulafungin is set. Working with the MEPAN Foundation, we’re in discussions with clinicians to see who will step up to the plate.
Looking further into the future, the results of this project open up the possibility of using GenAI and medicinal chemistry to synthesize of a library of anidulafungin derivatives in search of a near neighbor with optimized MECR pharmacological chaperone activity. We’ve already had preliminary discussions with Prof. Scott Lokey who specializes in cyclic peptides. Echinocandins are 6-mer cyclic peptides, so there are 64 million (20^6) possible total combinations to explore.
GenAI can perform an in silico screen to narrow that vast chemical space to 100-1,000 cyclic peptides that can be empirically assessed using reinforcement learning from yeast, fibroblast and fly feedback. We can also try to dial in oral bioavailability so that the we don’t need IV administration, which is how the echinocandins are currently administered to patients.
We still have to cross the threshold of the clinic but after several years of collaboration and research, we’re almost there. Onward!