SELENON Cure Odyssey
A CureMap prepared for Madeline Hollander & Sam Parker on behalf of their son Maxfield and other families affected by SELENON-related myopathy.


Executive Summary
The goal of the SELENON CureMap is to stimulate the development of medicines that will treat SELENON-related myopathy (SELENON-RM). A 6-stage CureMap outlined below is our vision of the path to a 1-to-N clinical trial for SELENON-RM. This assessment will focus on the first four stages: (1) model generation; (2) assay development; (3) compound screening; (4) confirmation of hits.
Based on the current state of knowledge on SELENON-RM, we recommend the development and characterization of several cellular disease models, and primarily pursuing drug repurposing given the slow progression of disease. We will also include an overview of the possibilities offered by AAV and mRNA therapy for this condition along with an overview of current knowledge of antioxidant treatment for SELENON-RM. Our key recommendations for the next 12-18 months are listed below.

Therapeutic track 1: Drug repurposing:
Stage 1: Pilot assay screens:
In collaboration with Dr. Alan Beggs, wild-type and knockdown C2C12 cells will be used in two pilot studies.
Stage 1A: Cell Painting: To identify a screenable phenotype to develop an assay to enable high-throughput screening of repurposed drugs. All eight dyes will be used in this assay to avoid missing a phenotype.
Stage 1B: R-Catcher ER+: To identify a screenable phenotype to develop an assay to enable validation of hits or a small screen consisting of hand-picked compounds.
Stage 2: Cell models: Identification of cell models with screenable disease phenotypes
Stage 2A: Fibroblasts: In collaboration with Charles River Laboratories or a Perlara popup cure lab, generate Maxfield’s fibroblast along with at least two other patient and three control fibroblasts lines. Test successful assays from stage 1A and stage 1B for a disease phenotype in fibroblasts.
Stage 2B: Myotubules: Utilizing Stan Nelson’s less invasive needle biopsy protocol patient-derived myoblasts to be generated for differentiation into myotubules. Test successful assays from stage 1A and stage 1B for a disease phenotype in myotubules.
Stage 3: Compound Screens: To screen compounds in stage 1 assays with stage 2 cell-models
Stage 3A: Pilot a compound library with ~2,500 compounds (Pharmakon) in collaboration with Charles River or a Perlara popup cure lab in chosen assay from stage 2.
Stage 3B: Scale up to a compound library of ~8,000 compounds (TargetMol) in assay chosen from stage 2.
Stage 3C: Hand-picked screen (optional): Include LAMA2 drug KH176 and glucose disposal drugs.
Stage 4: Confirmation of hits:
Stage 4A: R-Catcher ER+: Choose top hits from compound screen to test in Yang lab with patient generated fibroblasts or myotubules (Stage 2a/Stage 2b) for intracellular calcium re-filling of the endoplasmic reticulum.
Stage 4B: Muscle-biopsy contraction: Choose top hits from compound screens to test in Zito lab with muscle biopsies for excitation-contraction coupling assay.
Stage 4A: Zebrafish (conditional and optional): Choose top hits from compound screens to test in the Beggs lab in zebrafish model (if genetic rescue if successful).
Brief overview of alternative therapeutic strategies:
Therapeutic Track 2: Adenovirus gene therapy
Permanent: Gene-specific delivery of a functional wild-type SEPN-1. Close collaboration with the Beggs lab.
Therapeutic Track 3: mRNA therapeutic gene delivery
Transient & localized: gene-specific delivery of a functional wild-type SEPN-1 to target location (diaphragm) and other most affected muscles.
Therapeutic Track 4: Antioxidants
N-acetylcysteine
Vincerinone (EPI-743)
Authors
Charlotte Dunne, PhD (Cure Guide Perlara, PBC)
Ethan O. Perlstein, PhD (CEO of Perlara PBC & Maggie’s Pearl LLC)
Reviewers
Dr. Alan Beggs (Director of Manton Center for Orphan Disease Research, Boston’s Children’s Hospital).
We invite further post-publication review of this CureMap here on Substack.
INTRODUCTION
Maxfield is a 1-year boy who was born with SELENON-related myopathy (SELENON-RM formerly known as SEPN-1), a congenital muscular disorder caused by a nucleic acid duplication resulting in a frameshift mutation c.713dup (p.Asn238Lysfs*63) in the SEPN1 gene. SELENON-RM are a group of congenital muscular disorders that are characterized by slowly progressive axial muscle weakness and fatigue leading to early-onset spinal rigidity, scoliosis and life-threatening respiratory failure (Filipe et al., 2021). SELENON-RM is caused by loss-of-function variants in the SELENON gene and/or the genes encoding the Sec machinery required for the normal selenoprotein N-1 function (Santesmasses & Gladyshev, 2021). The prevalence of SEPN-1 related SELENON myopathy is estimated to be 0.5 in 1,000,000 (Witting et al., 2017). In addition to a homozygous pathogenic mutation in the SELENON gene, Maxfield also has heterozygous pathogenic mutation in the FKRP gene, a heterozygous mutation of unknown significance in the GYS1 gene and a heterozygous mutation in the MYO18B gene also of unknown significance. Although a heterozygous mutation in the FKRP gene was listed as pathogenic in Maxfield’s report, generally muscular dystrophy associated with FKRP mutations is autosomally recessive meaning that for penetrance of the disease, Maxfield would need to be homozygous for the mutation and not heterozygous. From the genetic report the homozygous mutation in the SELENON protein is most likely the necessary and sufficient driver of Maxfield’s symptoms and so this report will focus on Selenon-related myopathies. The contribution of these other genetic mutations will be discussed in more detail in regard to their potential role as disease modifiers.
SELENON - Gene
The SELENON gene encoding Selenoprotein N-1 is located on chromosome 1p36. It contains 13 coding exons (Moghadaszadeh et al., 2001) and two alternatively spliced variants. SELENON is a large gene with a transcript length of 4,212 base pairs. Translation of the SELENON gene results in a 590 amino acid length protein, Selenoprotein N-1. Selenoproteins are any proteins which contain a selenocysteine (Sec, U or Se-Cys) amino acid residue. Selenocysteine is an analog of the more common amino acid cysteine, with selenium attached to it. Selenium is an essential micronutrient with important functions in human health. Selenocysteine requires special machinery for insertion into the protein during translation. The 3’ untranslated region (UTR) of the Selenoproteins have a unique stem-loop structure in common which is required for selenocysteine (Sec) insertion sequence (SECIS). The 3’ UTR stem-loop structure recognises a UGA codon within the SELENON gene which, under ordinary circumstances, is recognized as a stop codon, but with the 3’ UTR is instead recognised as a Sec insertion codon. A second stop-codon redefinition element (SRE) next to the UGA codon has been identified as a conserved structure which stimulates readthrough at the UGA Sec insertion codon and augments the insertion efficiency of Sec. The importance of Sec insertion into Selenoproteins is to give them oxidative-reductase function. All selenoproteins have at least one Sec amino acid but can contain multiple Sec amino acids.
Selenoprotein N-1 - Protein
Selenoprotein N-1 has 590 amino acids of which the 21st amino acid is the Sec insertion site. There are 25 selenoproteins encoded in the human genome, and their synthesis requires a dedicated machinery. Mutations in this synthesis machinery result in SELENON related myopathies, giving further evidence to loss-of-function of SELENON in SELENON-related myopathies. Selenoprotein N-1 is an endoplasmic reticulum transmembrane glycoprotein. It contains an EF-hand domain which is used to sense the calcium concentration within the endoplasmic reticulum. Selenoprotein N-1 also contains a thioredoxin reductase-like domain on its endoplasmic reticulum side required for its role in regulation of oxidative stress.
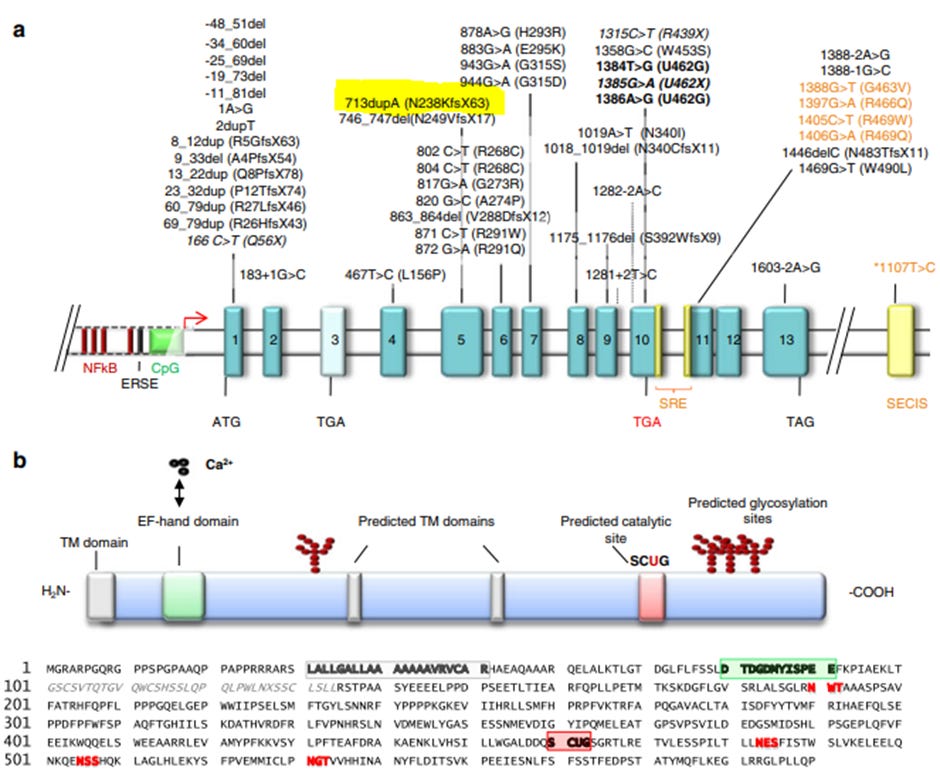
Figure 2. SEPN1 gene and Selenoprotein N-1 in humans. A) Dashed lines indicate an untranscribed region of the gene, upstream of the start-site (red arrow). Exons are depicted in blue boxes, exon 3 in light blue is alternatively spliced. Exon 5 contains the frameshift mutation 713dupA which is the same mutation that Maxfield has. In orange, the SECIS sequence within the 3’UTR and the SRE sequence are shown along with mutations found in these regions. B) Selenoprotein N-1 contains a transmembrane domain shown in gray, an EF-hand domain in green, a catalytic site in red and glycosylation motifs in bold red letters. Image is from (Castets et al., 2012).
Gene mutation - SELENON
Maxfield carries a homozygous mutation in SELENON at c.713dup (p.Asn238Lysfs*63) which can also be written as (N238KfsX63) but means the same. At the 713th nucleic acid base pair, a cytosine (C) is duplicated resulting in a frameshift mutation that adds an additional 63 nonsense amino acids to the first 238 amino acids of the protein before reaching a new stop codon produced by the frameshift mutation that results in a truncated Selenoprotein N-1. This mutation has been described in multiple patients (Arbogast et al., 2009; Ferreiro et al., 2002; Filipe et al., 2021). The relationship between genotype (specific mutations) and phenotype (manifestation of symptoms) is not clearly defined for Selenoprotein N-1 however most mutations in Selenoprotein N-1 as well as mutations in machinery required for Selenocysteine insertion produce a phenotype which overlaps with that of patients with mutations in the ryanodine receptor 1 (RyR1) calcium channel and in LAMA2 (Bouman et al., 2021; Jurynec et al., 2008). Although the phenotypes overlap, the spectrum of clinical features is heterogeneous. The most common feature of these mutations is spinal rigidity and respiratory impairment due to weakness in the diaphragm muscle. The main histopathological feature of Selenoprotein-related myopathy are minicores - areas of mitochondrial depletion and sarcomere disorganization.
Functions - Selenoprotein N-1
The known functions of Selenoprotein N-1 include calcium sensing, modulation of calcium entry and exit of the ER through maintenance of the oxidation states of calcium pumps responsible for the entry of calcium (SERCA) and exit of calcium (RyR) from the ER, and, more recently, the role of Selenoprotein in the association of mitochondria with endoplasmic and sarcoplasmic reticulum for ATP energy exchange.
A. Calcium sensing
The EF-hand motif is responsible for calcium sensing activity of Selenoprotein N-1. The double-helix structure of the EF-hand motif binds to calcium. Calcium is stored within the endoplasmic reticulum/sarcoplasmic reticulum and upon excitation calcium is released from these stores to initiate contraction signaling. Following an excitation-contraction coupled event, calcium stores are low within the sarcoplasmic reticulum, Selenoproetin N-1 through the EF-hand motif recognises low calcium levels and is activated. Selenoprotien-1 is involved in modulation of calcium signaling in both the endoplasmic reticulum and the mitochondria (Fig. 3). In its activated state, Selenoprotein N-1 modulates the calcium reuptake pump.

Figure 3. Selenoprotein N-1 regulates ER-mitochondrial calcium homeostasis. Measurements of [Ca2+] using recombinant aequorin-based Ca2+ sensors in the mitochondria upon agonist stimulation (100 μM Histamine) (N = 10). SEPN1-deficient cells had less ER Ca2+ (Fig. 6a) and transferred less Ca2+ to mitochondria (Fig. 6b). Reintroduction of SEPN1 (with the native selenocysteine) improved calcium accumulation in the ER and calcium transfer to the mitochondria, contrary to two SEPN1 mutants [in which either the selenocysteine was mutated into the similar amino acid cysteine (SEPN1 CC), or the couple of redox amino acids CU were mutated in two serines (SEPN1 SS)] (Filipe et al., 2021).
B. Modulation of calcium reuptake pump
When calcium is no longer binding to the EF-hand motif of Selenoprotein N-1 during calcium depletion following excitation, the conformation of Selenoprotein N-1 changes, likely exposing the selenocysteine more. Exposure of the selenocysteine facilitates the redox reduction of the sarcoplasmic/endoplasmic reticulum calcium (SERCA) pump (Fig. 4). When the SERCA pump is oxidized it is inactive. Upon reduction through Selenoprotein N-1 activation, SERCA is activated to pump calcium from the cytosol back into the sarcoplasmic/endoplasmic reticulum. Similar to the interaction with SERCA, Selenoprotein N-1 modulates the Ryanodine R1 (RyR1) calcium channel. The modulation of RyR1 could explain why mutations in both SELENON and RyR result in diseases with overlapping phenotypes (Jurynec et al., 2008).

Figure 4. SEPN-1 Calcium-handling in the sarcoplasmic reticulum. SEPN-1 senses calcium levels within the sarcoplasmic reticulum by its EF/hand motif, when calcium levels are low SEPN-1 activates the SERCA calcium pump through a redox reaction involving the thioredoxin-like domain (CU). Calcium (blue circles) is taken up into the sarcoplasmic reticulum by the calcium pump SERCA in its reduced state. When SERCA is oxidized it no longer takes calcium up. Calcium is stored inside the sarcoplasmic reticulum and is utilized in excitation-contraction coupling of skeletal muscles. Upon excitation, the ryanodine receptor 1 (RyR1) calcium channel is activated through dihydropyridine receptor (DHPR) channel located on the t-tubule, to release calcium in order to initiate muscle contraction. Image from (Zito & Ferreiro, 2021).
C. Mitochondrial associated membranes (MAMs)
A more recently discovered role of Selenoprotein N-1 is the anchoring of mitochondrial membranes to the endoplasmic reticulum to allow for calcium and ATP exchange between the two organelles. Selenoprotein N-1 is enriched at mitochondria-associated membranes (MAMs) in both muscle biopsies and in vitro models of Hela and C1212 cells. Selenoprotein N-1 is required for the integrity of ER-mitochondria contacts and is needed for calcium transients between the ER and mitochondria. In skeletal muscle, loss of Selenoprotein N-1 results in focal areas of mitochondrial depletion. In biopsies from patients with mutations in SELENON, impaired ER-mitochondria contacts and low ATP levels are demonstrated. In both in vitro and in vivo models, super resolution microscopy demonstrates a reduction in co-localisation of ER and mitochondria (Fig 5.). Induction of ER stress with the antibiotic tunicamycin (Tm) increases the co-localisation of ER and mitochondria but does not recover the mean branch length of the mitochondria network, which is reduced in Selenoprotein N-1 KO Hela cells (Fig 5. D). The relationship of ER and mitochondria is likely affected in SELENON models where co-localisation of ER and mitochondria is reduced and subsequent calcium transfer to mitochondria from the ER is reduced.
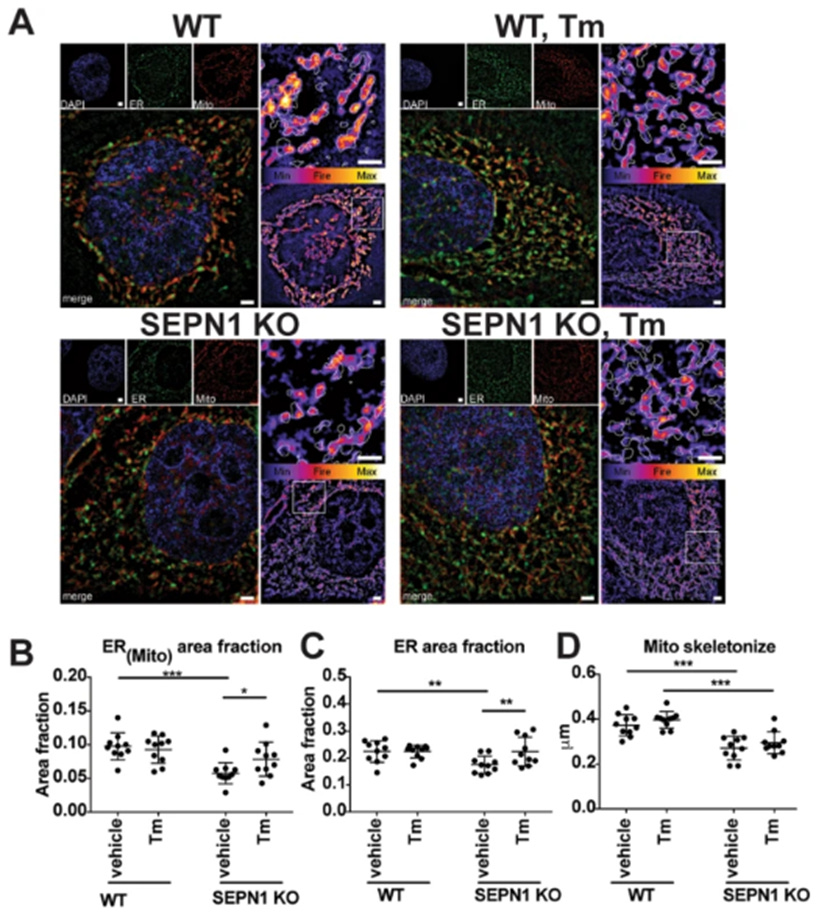
Figure 5. Co-localization study demonstrating higher co-localisation of ER and mitochondria in wild-type Hela cells (A). The fire inset image demonstrates the area of mitochondria found within the area of the ER mask (white dotted line). In the knockout (KO) Hela line, co-localisation of ER and mitochondria are reduced (A & B). From (Filipe et al., 2021).
Potential disease modifying gene mutations
From Maxfield’s genetic report, the homozygous pathogenic mutation in the SELENON gene is most likely the cause of his symptoms. Within the report there are three other genes one of which contained a pathogenic mutation in the gene encoding Fukutin related protein (FKPR) but normally only manifests in symptoms when a patient has two copies of this mutation. Although the single mutation in FKPR is solely not likely to cause the symptoms Maxfield is experiencing, the single mutation could be a modifier of the effects of the homozygous mutation in SELENON. Similarly to FKPR, two other mutations in Maxfield’s genetic report were listed as unknown significance, Glycogen Synthase 1 (GYS1) and Myosin XIIIB (MYO18B), they may also contribute through modification of the mechanism of loss of Selenoprotein N-1. Here we will examine these three genes and their possible role in modifying the mechanism of Selenoprotein N1-related myopathy with special regard to possible interference with mitochondrial to endoplasmic reticulum interactions, calcium refilling of the endoplasmic reticulum and thiol-redox activity in order to fully assess these three mechanisms as therapeutic screening strategies.
Fukutin related protein (FKPR)
Maxfield has a heterozygous mutation in FKPR, c.826C>A (p.Leu276Ile). Mutations in this gene have been associated with limb-girdle muscular dystrophy-dystroglycanopathy, type C5 (MDDGC5) which is caused by the NM_001039885.3:c.826C>A - missense variant (NCBI). It is usually an autosomal recessive disorder, so children carry two copies of the mutated form, and their parents who carry one copy are unaffected. Maxfield is heterozygous for the pathogenic variant. Limb-girdle dystrophy is a group of disorders that cause weakness and muscle wasting in the arms and legs. The muscles most affected are those closest to the body (the proximal muscles) more specifically the muscles of the shoulders, upper arms, pelvic area and thighs (Medline). Since Maxfield is heterozygous for the mutation in FKPR it is unlikely to be the cause of his symptoms although in rare cases it is possible for a heterozygous mutation to be causative of limb-girdle muscular dystrophy. Moreover, Maxfield’s muscle weakness is more prominent in his trunk and his diaphragm which are not muscles ordinarily affected in limb-girdle muscular dystrophy.
Muscle biopsies of patients with mutations in the FKPR gene demonstrate a reduction in laminin alpha 2 protein and a profound depletion of alpha-dystroglycan (Topalogu et al., 2003). Lamin alpha 2 is an extracellular matrix protein thought to be required for cell attachment, migration and organization within a tissue. Alpha-dystroglycan is a peripheral membrane component found in muscle, nerve, heart and brain, it is involved in bridging the cell membrane to the extracellular matrix. FKPR encodes a protein which is targeted to the golgi apparatus and is involved in the post translational modification of alpha-dystroglycan. Post-translational modification in the golgi apparatus often involves glycosylation. In the post translational modification of alpha-dystroglycan, a mutation in FKPR resulted in a profound loss of alpha-dystroglycan. Moreover, aberrant glycosylation of alpha-dystroglycan is suggested to be the primary cause of some congenital muscular dystrophies (Endo, 2005). Although a mutation in FKPR, a protein found in the golgi results in a loss of alpha-dystroglycan, it has not yet been proven that this is due to loss of glycosylation of alpha-dystroglycan but it is a plausible explanation especially since aberrant glycosylation of alpha-dystroglycan results in congenital muscular dystrophy. In line with this logic it is possible that FKPR is involved in the glycosylation of another glycosylated protein, Selenoprotein N-1. The predicted glycosylation sites of Selenoprotein N-1 are at positions ASN156, ASN449, ASN471 and ASN497 (Chernorudskiy et al., 2020). Maxfield’s gene mutation in SELENON results in a truncated Selenoprotein N-1 containing the first 238 correct amino acids and 63 additional amino acids. In this truncated protein only the first predicted glycosylation site ASN156 is present, the remaining 3 predicted glycosylation sites are missing as a result of Maxfield’s mutation. However if the glycosylation function of FKPR is altered due to the heterozygous mutation in FKPR that Maxfield is carrying, it is possible that glycosylation of the truncated Selenoprotein N-1 is altered too.
Glycogen synthase 1 - GYS1
Maxfield has a heterozygous mutation in GYS1, c.1763G>A (p.Arg588His). Mutations in this gene are associated with muscle glycogen storage disease. The gene GYS1 encodes an enzyme glycogen synthase 1 which is responsible for the production of glycogen in heart and skeletal muscles. Mutations in GYS1 can result in complete loss of glycogen in muscles. Glycogen is a major source of stored energy in muscle. A homozygous mutation in GYS1 can result in cardiac death in children, heterozygous carriers are generally unaffected (Cameron et al., 2009).
At the molecular level, the cause of death of the child carrying a homozygous mutation was thought to be due to metabolic complications of mitochondrial myopathy but upon further investigation was found to be resultant from anomalies indicative of mitochondrial proliferation and oxidative compensation (Cameron et al., 2009). A muscle autopsy showed a decrease in all of the components of the mitochondrial respiratory chain with an increase in the activity of citrate synthase. The authors of the paper interpreted these results as autolysis of the muscle cells due to the lack of glycogen. In SEPN-1 knockout mice, glycogen levels are unaffected compared to control mice quadricep muscles (Rederstorff et al., 2011). However in another SEPN-1 knockout mouse, following exercise the mice had lower blood glucose levels and faster depletion of muscle and liver glycogen (Filipe et al., 2021). If loss of Selenoprotein N-1 results in faster depletion of glycogen stores and a mutation in GYS1 also affects the production of glycogen, these effects could be additive.
If any cardiac abnormalities do arise for Maxfield, GYS1 should be further investigated; however with current information showing a homozygous mutation results in complications and heterozygous carriers are unaffected, GYS1 is not currently a main area of focus for Maxfield.
Myosin XIIIB - MYO18B
Maxfield has a heterozygous mutation in MYO18B, c.3968G>T (p.Gly1323Val). When in the nucleus, MYO18B can regulate muscle-specific genes whereas when MYO18B is in the cytoplasm it may regulate intracellular trafficking. The encoded protein, myosin XIIIB functions as a homodimer which may interact with F-actin. Mutations in this gene are associated with lung cancer. A novel mutation in MYO18B resulting in generation of a truncated protein was found to cause congenital myopathy in a Swiss patient (Mihaylova et al., 2020). The reported patient had delayed motor development but was able to walk by the age of 1.5 years and reported slow progressive muscle degeneration at the age of 47 years. Maxfield’s mutation in MYO18B mutation is c.3968G>T (p.Gly1323Val), and the mutation is located within the motor domain of Myo18B.
The role of MYO18B is known to be sarcomere assembly. The MYO18B protein is located on the Z-lines of skeletal and cardiac muscle on F-actin. Mutations in MYO18B resulting in muscle weakness demonstrated loss of normal banding indicating loss of thick filaments (Gurung et al., 2017). The functions of MYO18B are thought to be regulation of muscle specific genes when localized in the nucleus and when localized in the cytoplasm, it may play a role in intracellular trafficking. It is unclear which muscle specific genes are regulated by MYO18B. It is possible that genes regulated by MYO18B interact with Selenoprotein N-1. The exact mechanism of intracellular trafficking disruption caused by mutations in MYO18B is unclear. It is also possible that intracellular trafficking disruption may affect mitochondrial behavior since mitochondria rely on intracellular trafficking to deliver ATP to different parts of the cell.
In order to rule out the possibility of disease modification by FKPR, GYS1 or MYO18B heterozygous gene mutations, Maxfield’s fibroblasts could be CRISPR corrected for each mutation generating three isogenic corrected pairs that are still homozygous for Maxfield’s SELENON mutation but with each heterozygous gene mutation individually corrected to wildtype. In a fourth line one of Maxfield’s SELENON mutations will be corrected to wildtype. Before carrying out CRISPR and isogenic controls it would be beneficial to reach out to experts in relation to each gene FKPR, GYS1 and MYO18B for a comment on how they see the potential interaction with Selenoprotein N-1, mitochondria and their gene of expertise.
Therapeutic tracks:
Potential therapeutic tracks for SELENON myopathy include drug repurposing, AAV gene therapy or mRNA therapy. Each of these will be outlined below with a more detailed overview of therapeutic track 1 - drug repurposing as this is our recommended track. Advancements are already underway in preclinical mouse studies for a gene therapy for SELENON-RM in Dr. Alan Beggs laboratory in Boston’s Children's hospital. A therapeutic track involving mRNA therapeutics is possible although no groups are working on this track for SELENON-RM as of yet. mRNA therapeutics are a novel area and although there is a lot of potential for mRNA in the treatment of rare genetic diseases, there is still a lot of work that needs to be done until mRNA therapies are ready to use for rare gene families. Drug repurposing screens on the other hand utilize thousands of small molecules which already have to some extent a safety profile and therefore provide a much faster route from the laboratory to the clinic. Moreover, the risks associated with gene therapy are higher and permanent while the use of mRNA therapy has a lower risk due to its transient short-lived effect. That said, mRNA as a therapy is relatively new and therefore advancing an mRNA therapeutic to a clinical setting would come with more challenges. Finally, the quality of life and life expectancy of SELENON-RM patients is better and higher than that of patients with other rare genetic diseases. It is the opinion of SELENON professionals in the field that the risk of gene therapy is much higher compared to the gain it would provide to a patient because quality of life and life expectancy are high in these patients to begin with. This means drug repurposing with compounds that already have safety profiles is currently the best available therapeutic track for SELENON myopathy since the risks and the time required to reach the clinic are lower. Although drug repurposing would provide the fastest, cheapest and safest track to the clinic, an mRNA approach could be considered as a very specific personalized therapeutic approach with a safer profile than gene therapy.
Therapeutic track 1: Drug repurposing (Recommended):
To carry out a drug repurposing screen, a high throughput screen and a disease model are required. Since the loss of function of Selenoprotein N-1 is assumed for all SELENON mutations but a genotype phenotype relationship has been hypothesized and not yet confirmed, the use of patient-derived cells is important to screen for hits which are relevant to Maxfield. Although the loss of function phenotype has not been fully confirmed, SELENON knockout and knockdown models are commonly used for SELENON research and should be used for initial assay development experiments since they should produce an exaggerated disease phenotype. For the later drug repurposing screen experiments, the inclusion of multiple patient-derived cells with SELENON mutations is recommended since additional patient lines would provide confidence in hits that come up in the screen. Furthermore, there are concerns that cellular health due to handling can interfere with mitochondrial/stress functions where the use of multiple patient and control lines would be beneficial.
The most difficult step is to find a screenable phenotype. Cytosolic calcium signaling can be screened in a high throughput manner by a Fura2 assay, but it would not be sensitive enough for detecting a clear readout in the levels of calcium in endoplasmic reticulum (Dr. Ester Zito). The use of a transfectable calcium sensor such as R-Catcher ER+ (Dr. Jenny Yang) could be sensitive enough to detect both refilling and release of calcium from the endoplasmic reticulum, however each condition needs to be imaged live for a short period of time, which is fast but not realistic for 12,000 compounds. The relationship between mitochondria and ER is a potential screenable phenotype. The co-localisation of ER and mitochondria can be difficult to screen with super resolution microscopy as it is time consuming for each individual sample, however a high throughput imaging pipeline examining cellular organelles including ER and mitochondria could overcome this time consuming factor. Since we are unsure of exactly what the screenable phenotype will be, the best course of action is to work with the Cell Painting kit to image and analyze up to 3,000 readouts in a high throughput manner to identify the most reliable disease-specific cellular phenotype. Since we already have prior knowledge about the mechanism of SELENON mutations, the number of dyes could be reduced to select only ER and mitochondrial dyes, reducing the workload required for this step. However, to remain unbiased and to include any potential readouts all 8 dyes should be included in the initial assay screening stages. These two assays should be piloted first in cell models commonly used in SELENON research such as C2C12 mouse myoblasts in wild-type versus SELENON knockout including a positive control to rescue a disease phenotype such as the ERO-1 inhibitor EN460 (too toxic for human use) in order to determine which assay would be useful for a compound screen in patient-derived cells.
Studies of Selenon myopathy have looked at both patient-derived fibroblasts, patient-derived myotubules and muscle biopsies. Excitation-coupling which is affected in Selenon myopathy can only be examined in muscle biopsies. The refilling and release of calcium from the sarcoplasmic reticulum can only be examined in patient-derived myotubules. This calcium regulation can also be examined in patient-derived fibroblasts looking at the endoplasmic reticulum instead of the sarcoplasmic reticulum, however fibroblast calcium studies have yielded un-reliable results due high variability in results accounted for by passage number and confluency of fibroblasts. Finally, mitochondria and ER localisation can be examined in both fibroblasts and myotubules. The generation of patient-derived fibroblasts is less invasive than the generation of myotubules from muscle biopsies but since patient-derived myotubules are required to reliably observe calcium phenotypes and recent advances have been made in less invasive needle biopsies (Stan Nelson) for the generation of myoblasts and subsequent myotubules, it is recommended that both fibroblasts and myotubules should be generated from Maxfield in the first stage of this project. If the R-Catcher ER+ assay in C2C12 knockout cells provides a disease phenotype, then R-Catcher ER+ should be used in the patient-derived myotubules. In the cell-painitng assay provides positive results in C2C12 knockout cells, both patient-derived fibroblasts and patient-derived myotubules should be screened with this assay.
Once a screenable phenotype assay(s) and a disease model(s) are established, a pilot screen of repurposed compounds followed by a larger compound screen or optionally a hand-picked compound screen can be carried out. The compounds within these screens are either approved by the FDA, EMA or have undergone significant safety profiling. If one of these compounds are a hit, the road to the clinic should be much shorter than if a newly generated compound were to be a hit.
The compounds which produce the best results should then be chosen for further testing. Since the number of compounds will be reduced, it may be possible at this stage to regroup with Dr. Jenny Yang to utilize the calcium sensor R-Cather ER+ to test the effects of the hit compounds on calcium re-filling and release from the endoplasmic reticulum in patient-derived myotubules (Reddish et al., 2017).
The final compound hits can be sent to Dr. Ester Zito for screening in patient-derived muscle biopsies for excitation-contraction coupling, although detection of disease phenotype will need to be confirmed before testing compound screen hits. It has already been agreed that a muscle biopsy should only be performed if there is already a reason for a surgery. Furthermore, recent advances in needle biopsies provide a less invasive route to generating patient-derived myotubules (Barthelemy et al., 2020). If there were an occasion for a muscle biopsy, Dr. Ester Zito should be contacted and arrangements should be made. A buffer is required at the surgery which can be made up at a Perlara popup lab according to Dr. Ester Zito’s protocol. The sample needs to be transferred to the buffer immediately and transported to Dr. Ester Zito’s lab. The effect of the buffer takes about 15 days so there is no need to deliver the sample in 24 hours but it should take no longer than two weeks in transit. Upon receipt of the biopsy Dr. Ester Zito can test the compound hits on the excitation-contraction response in the patient biopsy. Alternatively, if there is no requirement for a surgery, efforts should be made to identify a patient with a similar mutation as that of Maxfield’s. A recent paper from Dr. Ana Ferriro’s group used a sample from a female with the same homozygous mutation as Maxfield (Filipe et al., 2021), perhaps this patient or another is willing to donate a muscle sample. In this case the facebook group for families of Selenon myopathy patients, identified by Madeline can be utilized, although researchers cannot enter the group, contact details for the administrators of the group have been provided.
Further confirmation of the compound hits can be carried out in Dr Alan Beggs lab with a SELENON knockdown zebrafish model. Prior work needs to be carried out in order to confirm that the knockdown can be genetically rescued in the zebrafish, if this work is successful the zebrafish model could also be utilized to test if our compound hits can rescue the disease-phenotype of spontaneous coiling of the zebrafish embryos in addition to other phenotypes identified by the Beggs lab.
The next stage would be to look at working with rodent models. Currently Selenon knockout models would be the best step forward. However, in 12-18 months there may have been advances in the field and there may be other options available. These options should be reviewed again at a later date.
Action Items:
Stage 1: Assay development
1A: Cell Painting to identify screenable phenotypes with a focus on two cellular compartments: the endoplasmic reticulum and the mitochondria to develop an assay to enable high-throughput screening of repurposed drugs.
Cell Painting involves high throughput analysis of 3,000 image-based readouts generated from images of fibroblasts stained with 8 cellular compartment dyes (Bray et al., 2016). Top parameters are chosen and further investigated. Alternatively, since there is strong evidence for the relationship between mitochondria and ER relationship to be affected in SELENON-derived cells, cell-painting can be carried out utilizing a fewer number of dyes to reduce costs and obtain a specified readout for the relationship between mitochondria and endoplasmic reticulum. A more unbiased approach would be to include all 8 dyes and all 3,000 readouts. Since the identification of disease phenotypes in SELENON-derived cells has been challenging for the past 20 years, it is recommended that all 8 dyes are included in the early pilot stages at least in order to retain as much information about the cells as possible.
1B: R-Catcher ER+ to identify disease phenotypes with a focus on calcium entry and exit in the ER/SR at the SERCA pump.
R-Catcher ER+ was a recently developed calcium channel tool which is specific for the ER and can be specifically localized to SERCA channels. This is a powerful advancement in calcium channel research and could prove to be significantly useful in detecting a SELENON disease phenotype in patient-derived myotubules. Initial pilot studies should be carried out with C2C12 mouse myoblasts in order to test R-Catcher ER+ as a tool to detect loss of function of SELENON in wildtype and knockout C2C12 cell lines.
Stage 2: Cellular Models
2A: Patient-derived fibroblasts: In collaboration with the Broad Institute or a perlara pop lab, generate Maxfield’s fibroblast along with at least two other patient and three control fibroblasts.
To ensure a reliable readout is obtained from the subsequent compound screen it is important to include more than one patient and control line. A minimum of three patient and control fibroblast lines each should be generated. More can be considered. Contract labs routinely generate patient fibroblasts, which can be transferred to another contract lab for subsequent compound screens. A less expensive alternative to a one-stop-shop CRO like Charles River is the Perlara popup lab network, which is staffed by scientists experienced in generating fibroblasts and running subsequent high throughput screens. Lines will be bulked up and frozen into 3-5 vials for long term cryo-storage for future use in various labs.
2B: Patient-derived myotubules: In collaboration with the Broad Institute, Perlara pop-up labs, the Beggs lab or Ferriro’s lab, from Maxfield and control’s less invasive needle muscle biopsy attempt to generate myoblasts and subsequent myotubules.
Stage 3: Compound Screens
3A: Pilot a compound library with ~2,300 compounds (UCSF Pharmakon library) in collaboration with Charles River or a Perlara popup lab in an assay chosen from stage 2.
UCSF Pharmakon comprimes ~2,300 compounds from the Microsource Spectrum and Prestwick collections. Plates of compounds will be screened in patient and control fibroblast lines to identify compounds which reduce the number of long range (100/200 nm) MAMs and increase the number of short range (20/30 nm) MAMs. Determining the number of short- and long-range MAMs will be carried out by cell-painting analysis chosen in stage 2.
3B: Scale up to a compound library of 12,000 compounds (Reframe) in assay chosen from stage 2.
Upon successful completion of stage 3A, stage 3B will commence with a larger compound library containing 12,000 compounds. Reframe is a best-in-class repurposing compound library containing nearly all small molecules which have reached clinical development or have undergone significant preclinical profiling. The benefit of using this compound library is it allows for rapid screening of compounds which already demonstrate a safety profile reducing the expensive research and development costs of trialing a new therapy (Janes et al., 2018).
3C: Hand-picked compound screen (Optional) for screening in the most reliable assay developed in this roadmap.
In order to test likely candidates, a list of compounds can be compiled which are approved or in testing for LAMA2 or other muscular diseases, in particular compounds which have antioxidant activity particularly thiol reducing.
Stage 4: Confirmation of hits
4A: ER calcium refilling: Choose top hits from compound screen to test in Yang lab with patient generated myotubules for intracellular calcium refilling of the endoplasmic reticulum.
Calcium refilling into the endoplasmic reticulum or sarcoplasmic reticulum (muscle cells) is defective in SELENON myopathy. Currently a general measurement of calcium in the cytoplasm is used to examine calcium signaling called Fura2 but this is not specific to calcium re-filling and is unlikely to be sensitive enough for the purpose of a compound repurposing screen. R-Cather ER+ is a calcium sensor which can be used to measure the release of calcium from the endoplasmic reticulum and the refilling of calcium back into the endoplasmic reticulum. This is a specialized approach which has not yet been used in the SELENON myopathy field, however Dr. Jenny Yang is an expert in calcium signaling and she has developed R-Cather ER+ which could be utilized in her group to test the phenotype of patient and control fibroblasts and to test the effect of the top hits from the compound screens from Stage 3A and Stage 3B on calcium refilling in patient and control myotubules.
4B: Excitation-contraction coupling: Choose top hits from compound screens to test in Zito lab with muscle biopsies for excitation-contraction coupling assay.
In order to avoid unnecessary invasive sampling, only if an operation is necessary for Maxfield, e.g some SELENON patients require surgery for spinal realignment, a muscle biopsy would be taken. Prior planning is required as the biopsy should be placed directly into a specific buffer for at least 15 days. Once in the buffer the biopsy should be shipped to Dr. Ester Zito. In Dr. Ester Zito’s lab, excitation-contraction coupling of the muscle biopsy can be tested. The top hits of the compound screens from Stage 3A, Stage 3B and Stage 4A should be tested in Dr. Ester Zito’s lab at this time. If Maxfield does not require a surgery, it is an option to take a muscle biopsy from the leg muscle. Alternatively, it is an option to ask older patients with SELENON if they would donate a muscle biopsy, preferably with the same mutation as Maxfield but since the genotype-phenotype relationship of SELENON mutations is not yet officially proven and all SELENON mutations seem to culminate in loss of function of the SELENON protein, biopsies could be tested from any patient with a SELENON myopathy who carries two mutated SELENON alleles.
4C: Zebrafish: Choose top hits from compound screens to test in Beggs lab with zebrafish SELENON model.
A zebrafish model for SELENON is being extensively characterized and developed in the Beggs lab, with a number of identified disease phenotypes available to test for rescue from hit compound treatment. Additional confirmation of disease the disease phenotype needs to be confirmed by genetic rescue prior to testing the hit compounds.
Therapeutic track 2: Gene Therapy
Dr. Alan Beggs of Boston’s children hospital is currently working on a gene therapy for SELENON-RM. Since loss of function of Selenoprotein N-1 is the overarching mechanism of action of all SELENON-RM patients, the gene therapy involves adeno-associated virus (AAV) delivery of the functional wild-type gene SELENON. The addition of a functional SELENON gene should be translated into a function Selenoprotein N-1 in patients which would compensate for the loss of their mutated Selenoprotein N-1. Although in vitro and in vivo data show promising results, the current barrier of this therapeutic track is finding biomarkers. Since this work is already underway in Dr. Alan Beggs lab, it may be a therapeutic option in the future. We recommend first exploring the drug repurposing track. If a gene therapy is developed and tested in clinical trials -- and proven to be well tolerated – it would be a terrific option for Maxfield because the main benefit of an AAV therapy is that only one dose is required. One dose of AAV integrates the gene of interest into the patient's genome and it would mean that no therapies are required. We advise to wait until this is an established therapy before trying it.
Therapeutic track 3: Messenger RNA Therapy (proving ground for irreversible AAV/CRISPR where the technology is rapidly evolving and improving)
Messenger RNA (mRNA) therapy is only recently widely accepted due to the coronavirus pandemic and its use as a vaccine therapy. Many advances have been made in designing mRNA therapies such as replacing all the uridine nucleic bases with pseudouridine to reduce the inflammatory response toward the mRNA therapy, altering the 5’ cap structure to modify transfection efficiency and altering the poly A tail to modify translation efficiency. In order to deliver the mRNA to a specific target, lipid nanoparticles are currently being developed. For the COVID-19 vaccine, lipid nanoparticles which when injected into the blood, target the liver were used but instead of injecting into the blood, the muscle was injected. Intramuscular injection of the liver lipid nanoparticle resulting in local delivery to the injected muscle.
A similar approach could be utilized for SELENON-RM where functional wild-type mRNA lipid nanoparticles (current liver formulations) are injected into muscles which are most affected in individual patients. This would provide a 1-2 week expression of functional Selenoprotein N-1 at a specific target muscle. The risk compared to gene therapy is reduced because the effect is transient, i.e. the mRNA is broken down and after two weeks there is no longer any protein expression from the therapy. If there were an adverse effect, therapy can simply be stopped. The risk of using an mRNA therapy is low but some people react to the lipid nanoparticles, this is true for the covid vaccine too, the risk is heightened in those who have previously received plastic surgery.
The barriers involved in developing an mRNA therapy include first cloning of a functional wilt-type selenoprotein into an in vitro transcription backbone. A challenge will arise here due to the unique nature of the 3’ UTR required for Sec insertion into the Selenoprotein N-1. Similar challenges will arise that the gene therapy track is currently facing in terms of biomarkers in animal models. Additionally, since the mRNA therapy is targeted to specific muscles, it may be more challenging to obtain FDA approval for intramuscular injection into different muscles. The therapy is novel and would require many in vivo animal studies which are notoriously quite costly in time and money. The clear benefit in a drug repurposing screen versus an mRNA therapy is the safety profile present or in part present for the compounds that will be screened.
Therapeutic track 4: Antioxidants
For the modulation of the calcium channels by Selenoprotein N-1, a redox reaction is required. The loss of Selenoprotein N-1 results in these calcium channels remaining in an oxidized state. One therapeutic avenue is to use antioxidants to drive the redox reduction of these oxidized proteins. Not all antioxidants will be beneficial for the treatment of Selenoprotein N-1. The main requirement is the ability of an antioxidant in thiol reduction to ensure the ability to modulate the SERCA2 calcium channel (Marino et al., 2014). N-Acetylcysteine has the ability to reduce thiol groups. A clinical trial was carried out in 7 Selenon patients which was completed in 2020 but the results of the study have not yet been posted (ClinicalTrials.gov). The use of antioxidants is advised while research is underway to identify a disease-modifying therapeutic.
We recommend a literature-based deeper dive to come up with rational drug repurposing candidates based on antioxidant mechanisms. Perlara can facilitate introductions to other families/foundations that have done rational literature-based drug repurposing in the mitochondrial disease space. Many have independently converged on Vincerinone (formerly EPI-743) as the most potent antioxidant but it is still in clinical development and only available via compassionate use at this time.
References
Arbogast, S., Beuvin, M., Fraysse, B., Zhou, H., Muntoni, F., & Ferreiro, A. (2009). Oxidative stress in SEPN1 -related myopathy: From pathophysiology to treatment. Annals of Neurology, 65(6), 677–686. https://doi.org/10.1002/ana.21644
Barthelemy F, Woods JD, Nieves-Rodriguez S, Douine ED, Wang R, Wanagat J, Miceli MC, Nelson SF. A well-tolerated core needle muscle biopsy process suitable for children and adults. Muscle Nerve. 2020 Dec;62(6):688-698. doi: 10.1002/mus.27041. Epub 2020 Sep 20. PMID: 32820569; PMCID: PMC7756388.
Bouman, K., Groothuis, J. T., Doorduin, J., van Alfen, N., Udink ten Cate, F. E. A., van den Heuvel, F. M. A., Nijveldt, R., van Tilburg, W. C. M., Buckens, S. C. F. M., Dittrich, A. T. M., Draaisma, J. M. T., Janssen, M. C. H., Kamsteeg, E.-J., van Kleef, E. S. B., Koene, S., Smeitink, J. A. M., Küsters, B., van Tienen, F. H. J., Smeets, H. J. M., … Voermans, N. C. (2021). Natural history, outcome measures and trial readiness in LAMA2-related muscular dystrophy and SELENON-related myopathy in children and adults: protocol of the LAST STRONG study. BMC Neurology, 21(1), 313. https://doi.org/10.1186/s12883-021-02336-z
Bray, M.-A., Singh, S., Han, H., Davis, C. T., Borgeson, B., Hartland, C., Kost-Alimova, M., Gustafsdottir, S. M., Gibson, C. C., & Carpenter, A. E. (2016). Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nature Protocols, 11(9), 1757–1774. https://doi.org/10.1038/nprot.2016.105
Cameron, J. M., Levandovskiy, V., MacKay, N., Utgikar, R., Ackerley, C., Chiasson, D., Halliday, W., Raiman, J., & Robinson, B. H. (2009). Identification of a novel mutation in GYS1 (muscle-specific glycogen synthase) resulting in sudden cardiac death, that is diagnosable from skin fibroblasts. Molecular Genetics and Metabolism, 98(4), 378–382. https://doi.org/10.1016/j.ymgme.2009.07.012
Castets, P., Lescure, A., Guicheney, P., & Allamand, V. (2012). Selenoprotein N in skeletal muscle: from diseases to function. Journal of Molecular Medicine, 90(10), 1095–1107. https://doi.org/10.1007/s00109-012-0896-x
Chernorudskiy, A., Varone, E., Colombo, S. F., Fumagalli, S., Cagnotto, A., Cattaneo, A., Briens, M., Baltzinger, M., Kuhn, L., Bachi, A., Berardi, A., Salmona, M., Musco, G., Borgese, N., Lescure, A., & Zito, E. (2020). Selenoprotein N is an endoplasmic reticulum calcium sensor that links luminal calcium levels to a redox activity. Proceedings of the National Academy of Sciences, 117(35), 21288–21298. https://doi.org/10.1073/pnas.2003847117
Endo, T. (2005). Aberrant glycosylation of alpha-dystroglycan and congenital muscular dystrophies. Acta Myologica : Myopathies and Cardiomyopathies : Official Journal of the Mediterranean Society of Myology, 24(2), 64–69.
Ferreiro, A., Quijano-Roy, S., Pichereau, C., Moghadaszadeh, B., Goemans, N., Bönnemann, C., Jungbluth, H., Straub, V., Villanova, M., Leroy, J.-P., Romero, N. B., Martin, J.-J., Muntoni, F., Voit, T., Estournet, B., Richard, P., Fardeau, M., & Guicheney, P. (2002). Mutations of the Selenoprotein N Gene, Which Is Implicated in Rigid Spine Muscular Dystrophy, Cause the Classical Phenotype of Multiminicore Disease: Reassessing the Nosology of Early-Onset Myopathies. The American Journal of Human Genetics, 71(4), 739–749. https://doi.org/10.1086/342719
Filipe, A., Chernorudskiy, A., Arbogast, S., Varone, E., Villar-Quiles, R.-N., Pozzer, D., Moulin, M., Fumagalli, S., Cabet, E., Dudhal, S., de Simoni, M.-G., Denis, R., Vadrot, N., Dill, C., Giovarelli, M., Szweda, L., de Palma, C., Pinton, P., Giorgi, C., … Ferreiro, A. (2021). Defective endoplasmic reticulum-mitochondria contacts and bioenergetics in SEPN1-related myopathy. Cell Death & Differentiation, 28(1), 123–138. https://doi.org/10.1038/s41418-020-0587-z
Gurung, R., Ono, Y., Baxendale, S., Lee, S. L. C., Moore, S., Calvert, M., & Ingham, P. W. (2017). A Zebrafish Model for a Human Myopathy Associated with Mutation of the Unconventional Myosin MYO18B. Genetics, 205(2), 725–735. https://doi.org/10.1534/genetics.116.192864
Janes, J., Young, M. E., Chen, E., Rogers, N. H., Burgstaller-Muehlbacher, S., Hughes, L. D., Love, M. S., Hull, M. v., Kuhen, K. L., Woods, A. K., Joseph, S. B., Petrassi, H. M., McNamara, C. W., Tremblay, M. S., Su, A. I., Schultz, P. G., & Chatterjee, A. K. (2018). The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proceedings of the National Academy of Sciences, 115(42), 10750–10755. https://doi.org/10.1073/pnas.1810137115
Jurynec, M. J., Xia, R., Mackrill, J. J., Gunther, D., Crawford, T., Flanigan, K. M., Abramson, J. J., Howard, M. T., & Grunwald, D. J. (2008). Selenoprotein N is required for ryanodine receptor calcium release channel activity in human and zebrafish muscle. Proceedings of the National Academy of Sciences, 105(34), 12485–12490. https://doi.org/10.1073/pnas.0806015105
Marino, M., Stoilova, T., Giorgi, C., Bachi, A., Cattaneo, A., Auricchio, A., Pinton, P., & Zito, E. (2015). SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Human Molecular Genetics, 24(7), 1843–1855. https://doi.org/10.1093/hmg/ddu602
Mihaylova, V., Chablais, F., Herenger, Y., Spiegel, R., & Heinrich Jung, H. (2020). Novel truncating mutations of MYO18B causing congenital myopathy in a Swiss patient. Neurology Genetics, 6(4), e458. https://doi.org/10.1212/NXG.0000000000000458
Moghadaszadeh, B., Petit, N., Jaillard, C., Brockington, M., Roy, S. Q., Merlini, L., Romero, N., Estournet, B., Desguerre, I., Chaigne, D., Muntoni, F., Topaloglu, H., & Guicheney, P. (2001). Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nature Genetics, 29(1), 17–18. https://doi.org/10.1038/ng713
Reddish, F. N., Miller, C. L., Gorkhali, R., & Yang, J. J. (2017). Monitoring ER/SR Calcium Release with the Targeted Ca<sup>2+</sup> Sensor CatchER<sup>+</sup> Journal of Visualized Experiments, 123. https://doi.org/10.3791/55822
Rederstorff, M., Castets, P., Arbogast, S., Lainé, J., Vassilopoulos, S., Beuvin, M., Dubourg, O., Vignaud, A., Ferry, A., Krol, A., Allamand, V., Guicheney, P., Ferreiro, A., & Lescure, A. (2011). Increased Muscle Stress-Sensitivity Induced by Selenoprotein N Inactivation in Mouse: A Mammalian Model for SEPN1-Related Myopathy. PLoS ONE, 6(8), e23094. https://doi.org/10.1371/journal.pone.0023094
Topaloglu, H., Brockington, M., Yuva, Y., Talim, B., Haliloglu, G., Blake, D., Torelli, S., Brown, S. C., & Muntoni, F. (2003). FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology, 60(6), 988–992. https://doi.org/10.1212/01.WNL.0000052996.14099.DC
Witting, N., Werlauff, U., Duno, M., & Vissing, J. (2017). Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark. Neurology Genetics, 3(2), e140. https://doi.org/10.1212/NXG.0000000000000140
Zito, E., & Ferreiro, A. (2021). Calcium and Redox Liaison: A Key Role of Selenoprotein N in Skeletal Muscle. Cells, 10(5), 1116. https://doi.org/10.3390/cells10051116