Shwachman-Diamond Syndrome/SDS Cure Roadmap
The first Cure Roadmap was commissioned in January 2021 by a pioneer parent named Julia Hawkins from the Shwachman Diamond Syndrome UK foundation, or SDSUK.

Thankfully, I was (re)introduced to Julia by Oli Rayner in October 2020. A YC founder had first introduced us in January 2019 as Perlara 1.0 was about to go off a cliff. I’m so glad we got reconnected because the timing was right. Julia and I worked diligently and diplomatically over the winter and spring of ‘21 seeking to forge a scientific consensus with community stakeholders including SDS Foundation, SDS Alliance and several EU country-specific groups. From the first drafts that were circulated in April and May 2021 to the SDSUK-hosted community Zoom convening on September 24, 2021, we incorporated wide-ranging perspectives from basic scientists to biotechnologists to parent advocates. This project made it possible for Perlara to reboot, and for that I’m forever grateful. And with that I present the SDS Cure Roadmap v1.

Shwachman-Diamond Syndrome/SDS Cure Roadmap
Prepared for SDSUK Foundation by Perlara PBC
October 2021

VISION
Toward a future where people with SDS live healthy, fulfilled and complete lives. This multi-year multi-modality drug development plan makes the scientific and commercial case for SDS. It calls for the creation of a $5M SDS Catalyst Fund, administered by SDSUK and Perlara in partnership with other SDS stakeholders, that will make strategic investments in therapeutics readiness, clinical trial readiness and SDS-focused biotech startups. This Roadmap envisions the following milestones and timelines:
In 2-3 years, precision oncology treatments informed by prognostic tests will be accessible to SDS patients of all ages; standardized natural history study data and regulatory agency-endorsed clinical trial protocols will be accessible to researchers, clinicians, companies and patients via federated data sharing platforms. Drug repurposing and clinical biomarker discovery enabled by SDS disease models and patient biosamples will augment the standard of care.
In 6-7 years, at least two new medicines that target the root cause of disease and reduce or prevent the onset of leukemia will be in clinical trials to treat 90% of SDS patients who have one or both of the two most common SBDS mutations.
In 10-12 years, the proceeds from a portfolio of live-saving prognostic tests and approved curative medicines made possible by the SDS Catalyst Fund will allow for the creation of a “long tail” fund that ensures all SDS patients around the globe get affordable access to these tests and medicines, including people living with SDS who have private SBDS mutations or non-SBDS gene mutations.

AUTHORS
Ethan O. Perlstein, PhD (CEO of Perlara PBC & Maggie’s Pearl LLC)
Julia Hawkins (SDSUK Trustee, SDS parent, General Partner at LocalGlobal and Latitude)
REVIEWERS
Professor Alan Warren, MD PhD (University of Cambridge)
Joan Mowery (President of SDS Foundation)
David Grainger, PhD (Chief Innovation Officer, Centessa Pharmaceuticals)
Yael Weiss, PhD (VP Business Development, Ultragenyx)
EXECUTIVE SUMMARY
The SDS Cure Roadmap takes the best of two complementary models of patient-driven drug development. Firstly, a mutation-agnostic approach that works for all patients regardless of gene mutation, as exemplified by the Cure SMA experience; and, secondly, a mutation-targeted approach that works for patients with a specific mutation whether it’s shared by many patients or just a few patients, as exemplified by the Cystic Fibrosis Foundation experience. A blended strategy is necessitated by the genetic architecture of SDS where two mutations in the gene SBDS are seen in 90% of SDS patients, while a “long tail” of 10% of SDS patients possess private SBDS mutations or mutations in a gene other than SBDS. For SDS patients flung onto this long tail of mutations, individualized treatment plans will be covered in a Roadmap addendum.
The SDS community is poised for capital-efficient, modality-diversified drug development. A SDS knowledge base has been forged by years of disease biology insights. A multi-species array of cell and animal SDS disease models already exist or can be quickly generated on a variant by variant basis. Multiple country-specific patient registries and patient-reported outcome measures have yielded possible clinical trial endpoints that will need to be formalized into a standardized clinical trial protocol, vetted by regulatory agencies and pressure-tested in single-patient “n-of-1” studies.
One drug target -- eIF6 -- has been robustly and reproducibly validated by both human and model organism genetics (Menne et al., 2007; Finch et al., 2011; Wong et al., 2011; Kennedy et al., 2021; Tan et al., 2021). Drug discovery efforts by a UK-based biotech company (SDS Therapeutics, Inc) are already underway to develop a mutation-agnostic novel oral drug that targets eIF6 in order to safely bypass the SBDS gene.
This Roadmap initiative was kicked off by Julia Hawkins and Ethan Perlstein in January 2021. During the spring months, we invited reviewers, families, and allied foundations to participate and have their voices heard. As a result, the Roadmap went through a transparent, rigorous and inclusive multi-month review process from April to September.
The goal of the SDS Cure Roadmap is to stimulate the formation of a $5M SDS Catalyst Fund that is initially housed within SDSUK and jointly operated with Perlara. This agile operating structure and a remote-first, globally distributed team will enable research capacity building and preclinical proof-of-concept studies across multiple programs that will coalesce into a biotech startup ecosystem urgently developing new life-saving prognostic tests and curative medicines.
The SDS Catalyst Fund will manage the $5M investment fund in order to deliver:
Precision oncology treatments for leukemia informed by single-cell genome sequencing
Unbiased drug repurposing using patient-derived cells (e.g., iPSCs) and animal models
Drafting of a standardized clinical trial protocol that will be validated by regulators in order to lower the barrier to investment into specific therapeutic tracks
Development of novel small molecules as disease-modifying eIF6 modulators, which would have utility for SBDS deficient patients as well as those on the long tail with EFL1 and DNAJC21 mutations
With respect to leukemia prevention, development of near curative mutation-targeted medicines that either correct or bypass one of the two common SBDS mutations in somatic (not germline) cells:
(i) a C-to-T base editor that corrects the 258+2 T→C mutation present in most SDS patients;
(ii) a splice-switching antisense oligonucleotide that relieves the splicing defect caused by the 258+2 T→C mutation;
(iii) a nonsense suppressor therapy (small molecule or tRNA modality) that reads through the premature stop codon created by the 183-184TA→CT mutation that is present in half of SDS patients.
Disease models, prognostic tests, clinical trial protocols and disease-modifying therapies developed for SDS may have broader applicability and impact across hematological cancers, inherited bone marrow failure syndromes, other monogenic ribosomopathies, longevity regulation and healthspan extension. This broader applicability will make investments into SDS compelling beyond the stakeholders who are directly affected by the disease.

INTRODUCTION
What is Shwachman-Diamond Syndrome?
SDS is a rare autosomal recessive disease with an incidence of approximately 1 in 80,000 live births. As points of reference: 1 in 2,000 live births is the threshold for a rare genetic disease and 1 in 50,000 to 100,000 live births is the threshold for an ultra-rare genetic disease. The first clinical reports of SDS described a disease that primarily affects bone marrow, pancreas and skeletal formation while also increasing life-time risk of hematological cancer in a subset of patients. Patients with SDS can present with failure to thrive early in life, neutropenia, short stature, and cognitive impairments.
The median age of survival is 41 years, with myelodysplastic syndrome (MDS), acute myeloid leukaemia (AML) and infection being the main causes of mortality. Approximately 30% of SDS patients develop life-threatening hematological cancer by age 30 (Donadieu et al., 2005). This urgent and dire need demands a plan of action on behalf of the 1,500-2,000 people living with SDS and their families in the USA and Europe – and the tens of thousands more living with SDS globally.
The involvement of multiple organ systems throughout development and into adulthood has led to palliative or disease-managing therapies that alleviate disease sequelae: pancreatic enzyme replacement therapy; fat soluble vitamin supplementation; stem cell growth factors; and bone marrow/hematopoietic stem cell transplantation. However, there are no approved disease-modifying medicines for SDS that target the root cause of disease.
Intriguingly, 90% of SDS patients would benefit from any one of several mutation-agnostic medicines that would generate cash flows that would be reinvested toward development of individualized therapies for the 10% of SDS patients on the long tail. This ability to step up where the market is hesitant has been exemplified by both Cure SMA and the Cystic Fibrosis Foundation. These groups are reinvesting proceeds from first-to-market medicines into curative treatment options for patients on the long tail.
Diversified portfolio of therapeutic modalities
The “90/10 split” genetic architecture necessitates a diversified portfolio of mutation-agnostic and mutation-targeted medicines that are embodied in distinct but combinable therapeutic modalities. Adding a layer of complexity, each SDS patient will require vigilant oncogene and tumor suppressor surveillance for leukemia-promoting somatic mutations (so called “double hits”) in patient bone marrow samples as well as assessment of rates of clonal expansion and variant allele frequencies.
Although country-specific registries and investigator-sponsored patient registries (e.g., North American SDS Patient Registry) have yielded valuable insights, a carefully crafted longitudinal natural history study is required in order to create comparator groups that will speed up clinical trials and render randomized placebo-controlled trial designs unnecessary. A network of clinical trialists is in place and different national SDS patient cohorts stand by. The UK SDS patient cohort in particular affords a strategic opportunity to leverage the UK centralized healthcare system, the genetic diversity of the UK population, and the burgeoning UK biotech startup ecosystem.
At the same time, standardized and published SDS disease models for the two most common SBDS variants are still needed. Specific disease models that will be enumerated below need to be created (or re-created) over the next 6-12 months and made accessible to any researcher or clinician or company that wants to work with them -- without restrictions. Once those models are in hand, the SDS Cure Roadmap contemplates advancing toward new medicines along four therapeutic tracks: (1) drug repurposing; (2) drug discovery; (3) gene/base editing; (4) nucleic acid therapies.
Ultimately, we predict that each SDS patient will require some form of combination therapy consisting of a mutation-agnostic medicine, e.g., small molecule eIF6 inhibitor that bypasses SBDS (and EFL1) gene function, and a mutation-targeted medicine, e.g., cytidine deaminase base editor that corrects the 258+2 T→C mutation. In a combination therapy scenario, an orally bioavailable small molecule medicine that reaches all cells of the body may be combined with another therapeutic modality that has restricted tissue tropism, or restricted tissue delivery, or is delivered to patient cells outside of the body. For example, a cytidine deaminase base editor delivered ex vivo to hematopoietic stem cells by lipid nanoparticles (like those used in the COVID-19 mRNA vaccines by Pfizer and Moderna) followed by reinfusion of edited cells back into the patient, as is current practice for autologous CAR-T immunotherapy.
Gene therapy needs de-risking
All therapeutic modalities are currently on the table except adeno-associated-vector-based gene therapy, which may be inherently challenging because a functional SBDS transgene would have to be safely delivered to all cells and tissues. Targeting potentially pre-cancerous cells with a viral vector modality which may integrate randomly into the genome is also a cause for concern. Finally, SBDS overexpression rescues disease phenotypes in a variety of SDS disease models but the long-term safety of chronic SBDS overexpression in humans presents significant unknown risks at this time.
In other words, there may be a narrower and much less forgiving clinical safety margin for systemic SBDS gene delivery approaches as compared to mutation-correcting medicines like sequence-specific base editors that may be delivered to hematological cell types ex vivo or safely targeted to specific organ systems in vivo. That said, targeted insertion into so-called safe harbors by lentiviral delivery, or a successor viral technology, is an avenue that deserves further attention but is not currently prioritized given the plethora of other therapeutic modality options. Given that risk, expensive multi-year preclinical de-risking studies in non-human primates will likely be required before any first-in-human studies are allowed to proceed.
Balancing mutation-agnostic and mutation-targeted approaches
This Roadmap draws inspiration from the Cure SMA case study. Basic research and preclinical collaborations supported by Cure SMA led to a key disease biology insight that put multiple therapeutic modalities in play: every SMA patient carries one or more silent backup copies of the damaged SMN1 gene called SMN2. Three different commercial sponsors gained regulatory approval for a mutation-agnostic curative medicine based on that ecosystem-enabling insight: a viral-vector-based SMN1 gene replacement therapy (Zolgensma®, Novartis); an antisense oligonucleotide that activates the SMN2 backup replacement gene (Spinraza®, Ionis/Biogen); and a small molecule drug that activates the SMN2 backup replacement gene (Evrysdi®, Roche).
However, unlike the Cure SMA experience which involved only mutation-agnostic medicines, SDS will require mutation-targeted approaches because there is no SBDS backup copy in the human genome. For that reason, the SDS Cure Roadmap also draws inspiration from the Cystic Fibrosis Foundation case study. A multi-decade venture philanthropy partnership with the biotech company Vertex led to three successive approvals for novel mutation-targeted small molecule potentiators and correctors that collectively treat 90% of the total CF patient population based on their specific CFTR mutations: Kalydeco® in 2012 initially for cystic fibrosis patients with the G155D mutation; Orkambi® in 2015 for cystic fibrosis patients with two ∆F508 mutations; and Trikafta® in 2019 for cystic fibrosis patients who have one ∆F508 mutation.
The aforementioned highly skewed genetic architecture of SDS is unique: the SBDS gene has dozens of known mutations but two common mutations are seen in 90% of all SDS patients regardless of genetic background. This situation is quite different from the cystic fibrosis example where the CFTR gene has over 1,700 (mostly long-tail) mutations and the prevalence of the most common mutation (“∆F508”) observed in CF patients varies depending on their genetic background.
The 90/10 skew in SDS genetic architecture results from a copy error during genome replication caused by the existence of a fossilized and partially deteriorated copy of SBDS inappropriately colliding with the SBDS gene sequence and leaving behind the 258+2 T→C mutation. The 90/10 skew actually narrows the therapeutic search space and creates strength in numbers from a drug commercialization standpoint.
The 258+2 T→C mutation can be targeted by multiple therapeutic modalities, as will be described in the sections below. The 258+2 T→C mutation results in trace amounts of full-length protein due to inefficient splicing, so in theory a therapeutic that potentiates SBDS activity could provide benefit. Similarly, private or rare SBDS missense mutations may be amenable to mutation-targeted small molecule potentiators or correctors.
As for the 183-184TA→CT frameshift mutation, it results in a truncated protein caused by a premature stop codon, and so small molecule chaperones, potentiators or correctors like the drugs developed for cystic fibrosis simply have little or no protein substrate to rescue. If ataluren is successful in a planned single-patient study in Italy under the direction of Dr Marco Cipolli, next-generation approaches that specifically read through the nonsense mutation downstream of the 183-184TA→CT would need to be developed, possibly in collaboration with PTC Therapeutics.
In analogy to the SMA oral drug Evrysdi, a novel oral small molecule that bypasses the essential ribosome-binding function of SBDS would have the broadest therapeutic application. eIF6 appears to be the primary target for a SBDS bypass based on: (1) deep mechanistic understanding of ribosome maturation; (2) empirical observation of somatic loss-of-function EIF6 mutations in SDS-patient-derived bone marrow stem cells; (3) ability of patient-derived EIF6 mutations to fully and effectively rescue the early lethality of a Drosophila SDS model hypomorphic for SBDS function (Kennedy et al., 2021; Tan et al., 2021).
Unbiased drug screens using a suite of new and previously published SDS disease models will likely yield additional drug targets beyond EIF6 that bypass SBDS essentiality. As SBDS may have secondary functions in addition to the final quality control step in ribosomal assembly, complete bypass of SBDS may only be possible in specific tissues or at specific times or in response to specific environments.
Balancing patients and profits
A patient-aligned model is appropriate to direct funds toward milestone-based sponsored research projects performed in academic or contract research labs that have contractually defined deliverables. The initial focus will be the generation of disease models and collection of patient tissues (skin biopsies, blood cells, bone marrow biopsies) for biobanking and establishment of induced pluripotent stem cell (iPSC) lines that will be made available to researchers via biorepositories. The SDS Catalyst Fund will advance a basic science discovery from an academic lab or a contract research lab to pivotal preclinical proof-of-concept studies be it in an animal model, a human iPSC-derived model or both.
When programs amass a compelling preclinical proof-of-concept data package, asset-centric early stage capital will be deployed to advance SDS therapeutic programs to a Phase 1/2a first-in-human clinical proof-of-concept study using a virtual startup creation model like RxCelerate that has been successfully deployed by the UK-based life science VC firm Medicxi. Once companies have been created, community alignment will occur by allowing the SDS Catalyst Fund to participate in commercial upside by receiving equity and/or stock options in SDS-focused startups in exchange for seed investment, pre-clinical proof-of-concept data packages, foundational IP and access to data from natural histories, patient registries, biomarker studies, disease modifier studies, and other sources. Cystic Fibrosis Foundation, among others, have been pioneers in patient alignment and community financial upside.
Moreover, SDS patient community members and foundations will be able to invest in syndicates alongside professional biotech investors, creating further alignment between patient community interests and potential biopharma partner interests. Proceeds from the sale or exit of those companies will be re-invested in realizing the full vision of this Roadmap, including investment in the 10% of SDS patients on the long tail. It is implicitly understood that this Roadmap is a living document subject to annual amendments and addenda in response to future technological developments.

THERAPEUTICS READINESS
The development of new medicines requires disease models, patient-derived biosamples and cells, and a deep mechanistic understanding of pathophysiology that identifies disease modifying drug targets, including the monogenic driver gene itself. The primary function of SBDS protein is in a fundamental biological process called ribosome licensing. Accordingly, simple cellular and animal models (patient cells, yeast, worms, slime mold and flies) can demonstrate very well the degree to which a given intervention normalizes, for example, a cellular phenotype such as polysome formation. Higher animal models then show how that translation deficit (either in quantity or quality) emerges as a phenotype, which varies between species and between tissues within a species.
But how much does the particular manifestation in, say, mouse bone marrow, tell us about humans? To the extent it’s the same as in humans, it’s probably also the same as in flies; and to the extent it differs from flies it is unlikely to be informative about humans. As a result, all information gained from disease models must be cross-validated and, as such, multiple models are needed to provide the best chance of therapeutic outcomes.
Disease models
Although half of SDS patients with SBDS mutations are compound heterozygous for the two recurring protein-truncating mutations, the 183-184TA→CT mutation in homozygosity is not observed in humans. Therefore, complete loss of SBDS is not compatible with life (Boocock et al., 2003). Given the historic challenges of creating a translationally relevant and viable mouse model of SDS, researchers and clinicians have studied human cell-based and small animal models over the last 20 years.
Although they are sometimes overlooked, invertebrate model organisms like yeast, slime mold, worms and flies benefit from facile genome engineering and the ability to create a suite of humanized patient avatars that are engineered to express any desired patient mutation. This is likely to be cheaper and more time efficient than the creation of large animal models. Also, invertebrate models have the added benefit of higher throughput, lower maintenance costs, and fast turnaround time.
Cell-based and animal models of SDS will be useful for a range of downstream applications. These uses include: (i) biomarker discovery using transcriptomics, proteomics, and metabolomics; (ii) further dissecting disease pathogenesis and pathophysiology in specific cell types or at specific developmental timepoints; (iii) disease modifier and genetic suppressor screens; (iv) high-throughput drug repurposing and drug discovery screens.
Below is description of the status of SDS disease models:
A humanized SBSD mouse model
In mice, unconditionally knocking out SBDS in all cells and tissues is incompatible with life (Zhang et al., 2006). Conditional mouse mutants where SBDS loss is spatially restricted to specific organs or cellular lineages have been generated but these models have limited utility beyond validation of certain classes of drug screening hit compounds.
We propose a singular focus on a humanized viable (i.e., hypomorphic) SDS mouse model that is on the critical path to the clinic. This model will be engineered to express the most common 258+2 T→C splice-site mutation harboring human-specific SBDS genomic sequence.
One of the main advantages of a humanized SBDS mouse is that mutation-targeted medicines like nucleic acid therapies or gene editing/base editor therapies can be tested directly in mice “as is” as clinical candidates rather than having to generate a human-specific clinical candidate and a murine counterpart molecule in parallel. The humanized SBDS mouse is currently being generated by Jackson Laboratory and will be made available to academic and industry researchers in the first half of 2022.
Cell-based models
Induced pluripotent stem cells, or iPSCs, are especially useful when a mouse model generation presents challenges. iPSC lines along with isogenic controls can be generated from peripheral blood mononuclear cells (PBMCs), which are collected via a blood draw, or from primary fibroblasts. SDS patient-derived iPSCs exhibit defects in endothelial, pancreatic and hematopoietic differentiation (Tulpule et al., 2013; Hamabata et al., 2020). These cellular phenotypes are therapeutically relevant when considering neutropenia and pancreatic insufficiency as primary outcome measures in any pediatric pivotal registration study.
Other cell types including human primary bone marrow stromal cells, lymphoblasts and skin fibroblasts exhibit disease phenotypes that could be adapted to a high-throughput image-based or flow cytometry-based primary drug screen (Austin et al., 2008; Wong et al., 2011; Tan et al., 2019). SDS patient-derived lymphoblast cell lines exhibit defects in ribosome assembly that would be a useful secondary assay in the validation of hits from other screens (Wong et al., 2011).
Zebrafish model
Zebrafish are excellent disease models for inborn bone marrow failures because of the ability to generate large experimental cohorts, optically transparent enabling real-time observation of blood cell differentiation, and quick development with all major organs having developed within the first five days. There is 86% sequence identity between human SBDS and its zebrafish ortholog. The SBDS ortholog was knocked down by morpholino in zebrafish to elucidate its role in development (Venkatasubramani & Mayer, 2008; Provost et al 2012). This resulted in defects in granulocyte migration and the development of the exocrine pancreas (with intact morphology of the endocrine pancreas), decreased neutrophil numbers with low migratory capacity, and bone and cartilage defects. Data from morpholino knockdown should always be considered provisional and possibly artifactual until confirmed by a bona fide genetic model.
Recently, CRISPR/Cas9 was used to introduce SBDS mutations in zebrafish (Oyarbide et al., 2020). Consequent impairment of ribosomal maturation was associated with the mutants phenocopying the clinical aspects of SDS patients with respect to reduced length and neutrophil numbers, and altered architecture of the intestinal folds, liver and pancreas. Pancreatic exocrine atrophy was supported by the reduction in lipid accumulation in mutants. Transcriptome analysis revealed upregulation of pro-apoptotic genes involved in the TP53 pathway and EIF6, and downregulation of ribosomal proteins. These mutants resembled starved wild type embryos, suggesting phenotypic dependency on malabsorption. The advantage of this model was survival through 21 days and in some cases, up to four weeks which marks the early juvenile phase.
Flies (Drosophila melanogaster)
Last year, a group created the first (and most simplified) SDS disease model in flies by knocking down the SBDS ortholog CG8549 in all neurons in the fly brain, which caused locomotive disabilities, mechanically induced seizures, hyperactivity, learning impairments, and anatomical defects in presynaptic terminals (Takai et al., 2020). The same limitations of morpholino knockdown models in zebrafish applies to RNAi knockdown models in flies.
A bona fide SDS fly model has been developed and characterized in Prof Alan Warren’s lab (Tan et al., 2021). As observed in other animals, reduced expression of the fly ortholog of SBDS results in lethality. Human SBDS functionally complements the Drosophila ortholog. Validating eIF6 as a therapeutic target, recurrent eIF6 mutations identified in primary SDS patient hematopoietic cells fully and effectively rescue the lethality of the hypomorphic SDS fly model.
Worms (Caenorhabditis elegans)
To date, no SDS bona fide worm model has been published. Deletion of the worm ortholog of SBDS is predicted to be lethal, as is the case for other species. Hypomorphic worm models with growth defects will be amenable to high-throughput drug and genetic suppressor screens.
Yeast (Saccharomyces cerevisiae)
Yeast knockout models of SDS have been instrumental in elucidating the precise role of SBDS and EFL1 in ribosome assembly and quality control (Menne et al., 2007). For the purposes of drug screening (both drug repurposing and drug discovery) and genetic suppressor screening, a knockout mutant of the yeast SBDS ortholog sdo1 exhibits a severe but suppressible growth defect. A temperature sensitive conditional Sdo1 mutant allele based on intein technology has also been generated (Tan et al., 2021).
Slime mold (Dictyostelium discoideum)
The slime mold Dictyostelium is another single-cell ancient eukaryote that can serve as a patient avatar both for drug screens and genetic suppressor screens (Wong et al., 2011; Tan et al., 2021).

CLINICAL TRIAL READINESS
In order to achieve the vision of curative medicines by the end of this decade, the 30% of SDS patients at highest risk of dying from MDS and leukemia will need to be identified using prognostic tests and other biomarker tests as early in the disease course as possible. Based on recent work by Prof Akiko Shimamura and colleagues showing that bone marrow stem cells that take a somatic mutation double hit to TP53 go on to become cancerous, while bone marrow cells that acquire protective loss-of-function mutations in EIF6 avoid a leukemic fate, we already know some of the targets to surveil in SDS patients (Kennedy et al., 2021). Somatic mutations in EIF6 rescue the fitness defect in SDS bone marrow stem and progenitor cells caused by the germline ribosome deficiency either by reducing the dose of eIF6 or by reducing eIF6 binding to the large ribosomal subunit (Kennedy et al., 2021; Tan et al., 2021).
Patient cohorts and clinical trialists are primed for single-patient observational safety and efficacy studies. As described below, a compassionate use (“n-of-1”) study of the repurposed drug ataluren will commence in Italy under the direction of Dr Marco Cipolli. That study protocol will serve as an initial impetus for standardization. However, the fragmentation of patient registries and biorepositories, as well as the siloing of data and biomaterials, needs to be overcome.
A global coalition of SDS foundations can unify existing retrospective national and investigator-sponsored natural history data onto a prospective digital platform. Pooling data from multiple registries and databases into a federated data sharing platform will enable real-time oncogenic surveillance to inform precision oncology treatments in time to make a difference. However, given the lack of standardization across country-specific registries, a new longitudinal, prospective (forward-looking) natural history study cohort will need to be assembled, as described below.
Data sharing
Fortunately, there is no need to reinvent the wheel in terms of data sharing platforms. The AT Children’s Project whole genome sequencing collaboration with the Broad Institute serves as a model for data sharing. RARE-X would make an excellent partner for a pilot project focused on SDS. Citizen, which was recently acquired by the genomics diagnostic company Invitae, is a digital patient registry platform and electronic medical records aggregator that could also be a potential collaborator. LunaDNA is a variation on the theme of a data sharing platform that gives patient communities ownership and commercial rights based on their shared data. Mission Bio is a cancer diagnostics offering single-cell hematology panels and could be the backend that powers any data collection effort.
Standardized clinical trial protocol
The SDS go-to-clinic strategy envisioned at this time is staggered: first a months-long, single-center pediatric pivotal registrational trial with a primary endpoint of improving blood counts (neutropenia/cytopenia) followed by a multi-year, multi-center pediatric and adult trials with a primary endpoint of leukemia prevention. A clinical trial protocol template will be developed based on those high level principles and will answer the following questions:
Is it all about leukemia risk, or how important is infection risk?
Is neutropenia or pancreatic insufficiency an endpoint in itself? Or a meaningful surrogate for early trials?
What about p53 mutated clones in bone marrow aspirates?
What about exploratory non-leukemia endpoints related to neurocognitive function?
Definitive natural history study
A clinical trial protocol template will massively reduce the barrier for investors coming into the SDS space if clinical trialists, and commercial sponsors of clinical trials, can point to how they are going to test their therapeutic candidate and that the primary outcome measure (or measures) are already accepted by regulators. A UK-cohort-based longitudinal natural history study to be conducted using a federated data sharing platform will allow for monitoring of the chosen or candidate endpoints over time, for example annually. That will create an invaluable dataset for clinical trialists around the world, showing the inter-patient and intra-patient variabilities in disease presentation they will need to impact in order to have their intervention considered a success by regulators. Even data over 2-3 years from 10-20 people could be a game changer.
If any one of the therapeutic tracks described below reaches the clinic over the next several years, there will be a demand of patients that could exceed availability. Maximizing efficiency will then be important, and there is much to be learned from the COVID-19 experience. For example, run a rolling cohort of standard-of-care through a clinical trial protocol and simply have companies/academics or whoever add comparator groups, rather than each having a separate randomized control trial (RCT) with its own control? This could be better than historical control, open-label studies yet no more costly or difficult to implement.
THERAPEUTIC TRACK 1: DRUG REPURPOSING
Drug repurposing is defined as identifying a new use for an already approved drug, be it a century old drug or a drug that was just recently approved. Drug repositioning is defined as salvaging an experimental drug that cleared the initial hurdle of early-stage safety studies but failed in late-stage efficacy studies for its intended disease indication.
The Broad Repurposing Hub (commercially available as the SPECS library) is an ideal launchpad for identifying drug repurposing and drug repositioning candidates. All validated SDS disease models will be screened against this collection as part of a coordinated multi-species high-throughput drug screening campaign.
Ataluren: go/no go
Based on a review of the biomedical literature, there is one target for which there is preliminary evidence for mutation-targeted drug repurposing: ataluren (Translarna®, PTC Therapeutics), which was approved by the EMA (but not yet the FDA) for the treatment of patients with Duchenne Muscular Dystrophy who have a nonsense mutation. Dr Marco Cipolli’s group in Italy showed that ataluren partially restored expression of SBDS protein and improved survival of multiple SDS patient derived cell types, including bone marrow stem cells that harbor the 183-184TA→CT mutation (Bezzerri et al., 2018). As noted above, Dr Cipolli’s group will be initiating a compassionate use, single-patient study in the coming months. While it is an approved drug in the EU, ataluren has failed clinical trials in the US for multiple disease indications.
Patient derived iPSCs in differentiation screens
A myeloid precursor differentiation assay using hematopoietic iPSC models and hit validation in zebrafish and mouse models was successfully used in drug repurposing screen for Blackfan-Diamond anemia (Doulatov et al., 2017). That case study serves as a model for how SDS patient derived iPSC-based lineage-specific differentiation screens will also be explored.
For example, there is support in the literature for conducting a high content, imaged-based screen for amylase expression in iPSCs induced toward pancreatic differentiation (Tulpule et al., 2013).
As a second example from that same study, a flow cytometry-based screen for expression of a differentiated myeloid cell marker (e.g., CD45) in iPSCs induced toward a hematopoietic fate would be feasible.
Patient derived primary cell lines in unbiased image-based screens
Downsides of iPSC-based screens are that they can take several weeks (or even months) of differentiation per screen and require considerable cell production capacity and lab operational resources. Therefore, a primary phenotypic screen using patient derived skin fibroblasts will be established as less expensive and more easily produced alternatives to iPSCs.
For example, SDS patient derived fibroblasts were first characterized over 15 years ago and exhibit an abnormal nucleolar morphology that would be compatible with an image-based high content screen (Austin et al., 2005).
The contract research organization Charles River Laboratories (CRL) is an attractive partner for these human cell-based drug screening activities. Proposals will also be solicited from other globally distributed CROs like WuXi and Evotec.
Model organisms in unbiased phenotypic screens
Complementing human cell models are small animal models, the fastest and cheapest of which is yeast. Several academic labs have published chemical and genetic suppressor screens using a sdo1∆ deletion mutant (Menne et al., 2007; Kanprasoet et al., 2015) or conditional temperature sensitive intein strains (Tan et al., 2021). According to an evolutionary pharmacology approach, primary drug screens will be performed using inexpensive invertebrates like yeast, worms and flies, reserving expensive and lower-throughput vertebrates like zebrafish or human differentiated iPSCs or patient-derived bone marrow aspirates for post-screening hit validation.
In fact, rescue of growth deficits of yeast and Dictyostelium SDS models could in principle serve as primary screening workhorses followed by hit validation in differentiated iPSCs, skipping larger animal models and bone marrow aspirates entirely. Assuming worm and fly SDS models can be made freely available to nonprofit and for-profit researchers alike, specialized biotech startups like Vivan Therapeutics and Modelis are equipped to perform drug repurposing screens for flies and worms, respectively.
THERAPEUTIC TRACK 2: DRUG DISCOVERY
The aforementioned existing and newly created (or recreated) SDS disease models will be pressure tested in the drug repurposing track. After they have demonstrated utility, these same disease models will be used in both mutation-agnostic and mutation-targeted drug discovery screens where novel chemical entities will be identified. While eIF6 constitutes the most validated drug target for SDS based on everything we know today, there may be equipotent SDS bypass suppressor pathways that have yet to be discovered.
Novel small molecule modulators of eIF6 binding to the ribosome
Perhaps the most compelling SDS drug discovery project already underway is a novel mutation-agnostic small molecule eIF6 binding modulator of eIF6 being incubated by Medicxi, a virtual biotech startup fund in the UK, and based on discoveries made in Professor Alan Warren’s lab at the University of Cambridge. Nomination of eIF6 as a bypass suppressor of SBDS comes from four independent but mutually corroborating lines of genetic evidence: (1) yeast lacking the SBDS ortholog SDO1 are sick and they acquire growth-restoring suppressor mutations in Tif6, the yeast ortholog of eIF6 (Menne et al., 2007); (2) the fitness defect of Dictyostelium cells lacking the SBDS homolog is rescued by expressing eIF6 mutations that have reduced affinity for the ribosome (Wong et al., 2011; Tan et al., 2021); (3) human bone marrow stem cell clones from SDS patients acquire suppressor mutations in eIF6 that have been interpreted as being potentially leukemia-protective (Kennedy et al., 2021; Tan et al., 2021); (4) expression of patient-derived eIF6 mutations in SBDS-deficient flies fully rescues the early larval lethality (Tan et al., 2021).
Novel 183-184TA→CT readthrough drug
An orthogonal, mutation-targeted drug discovery approach involves identifying an existing or novel ataluren analog, or a completely novel chemotype, to read-through the premature stop codon engendered by the 183-184TA→CT frameshift mutation (Bezzerri et al., 2020). The 183-184TA→CT frameshift mutation results in a completely non-functional protein, so even a small amount of readthrough culminating in full-length protein is predicted to have therapeutic benefit. This approach will gain considerable steam if clinical proof-of-concept data for ataluren are positive. What’s more, the 183-184TA→CT frameshift mutation can be targeted by multiple modalities, not just small molecules (see Track 4 below). PTC Therapeutics is a logical potential industry sponsor for this program.
Other targets beyond eIF6?
Are there other therapeutic mechanisms that suppress the loss or essentiality of SBDS? SBDS and EFL1 form a complex that evicts eIF6 (Weis et al., 2015). So what about a novel small molecule activator of EFL1, which may or may not provide mutation-agnostic therapeutic benefit?
And what about a novel small molecule activator of SBDS itself in cases where a sub-threshold amount of functional SBDS protein is present in cells? The genius of unbiased drug screens and integrating insights from multiple disease models throughout the drug screening process is that small molecule activators of EFL1 or SBDS could emerge as mechanistic classes.
The same disease models used to find novel small molecules can be reused again later for mechanism of action and target identification studies. Certainly for the SDS genetic long tail, screens will be designed to find small molecule potentiators or correctors specific for SBDS missense variants, as was done for CFTR missense variants in cystic fibrosis.
THERAPEUTIC TRACK 3: GENE/BASE EDITING
Patient derived cell models and the humanized SBDS mouse model will be used for testing an experimental mutation-targeted cytidine deaminase base editor designed to correct the 258+2 T→C single nucleotide polymorphism. The T→C substitution at position 258+2 disrupts an intronic donor splice site, resulting in mostly non-functional protein. However, the absence of suitable protospacer adjacent motif (PAM) sites near the 258+2T→C mutation will require a re-engineered base editor.
Much like in CAR-T, an SDS patient’s bone marrow stem cells can be mixed with a base editor ex vivo and then the correctly edited cells are infused back into the patient, limiting off-target effects that would be associated with systemic administration.
Furthermore, it’s possible that the selective growth and differentiation advantage of correctly edited hematopoietic stem cells means the on-target efficiency of base editing need not be 100%. The laboratory of Prof. Christian Brendel at Boston Children’s Hospital would be a sensible location for this project in collaboration with Prof. Akiko Shimamura’s group.
Base-editing amenable private mutations will require bespoke base editors. The team at Beam Therapeutics will be engaged for interest, as well as academic labs (e.g., Prof David Liu at Harvard) interested in working in this area.
THERAPEUTIC TRACK 4: NUCLEIC ACIDS
Splice-modulating ASO targeting 258+2 T→C
As a backup to the base editor approach described above in Track 3, a splice-modulating ASO based on the backbone chemistry and modifications of Spinraza (nusinersen) that sterically blocks the cryptic splice site favored in the wake of the 258+2 T→C mutation will be developed. The laboratory of Prof. Akiko Shimamura would be a sensible location for this project, and efforts toward this end are already underway.
Suppressor tRNA therapy targeting 183-184TA→CT
As a hedge to the small molecule nonsense readthrough modality, a AAV-delivered tRNA nonsense suppressor targeting the premature stop codon downstream of the 183-184TA→CT mutation will be developed in collaboration either with an academic lab or a biotech startup. A version of this technology called anticodon engineered transfer RNAs (ACE-tRNA) was developed to preclinical proof-of-concept with support from the Cystic Fibrosis Foundation (Lueke et al., 2019), which has its own long tail of nonsense mutations to contend with.
The three top tRNA biotech companies -- Tevard Biosciences, ReCode Therapeutics and Shape Therapeutics -- will be engaged for interest in a preclinical proof-of-concept study of full-length protein production across multiple patient-derived cell types.
Private ASO-amenable mutations
SBDS and non-SBDS intronic splicing mutations are extremely rare and will require special attention in the long tail addendum. n-Lorem Foundation is one option available today.
FUNDING MODEL
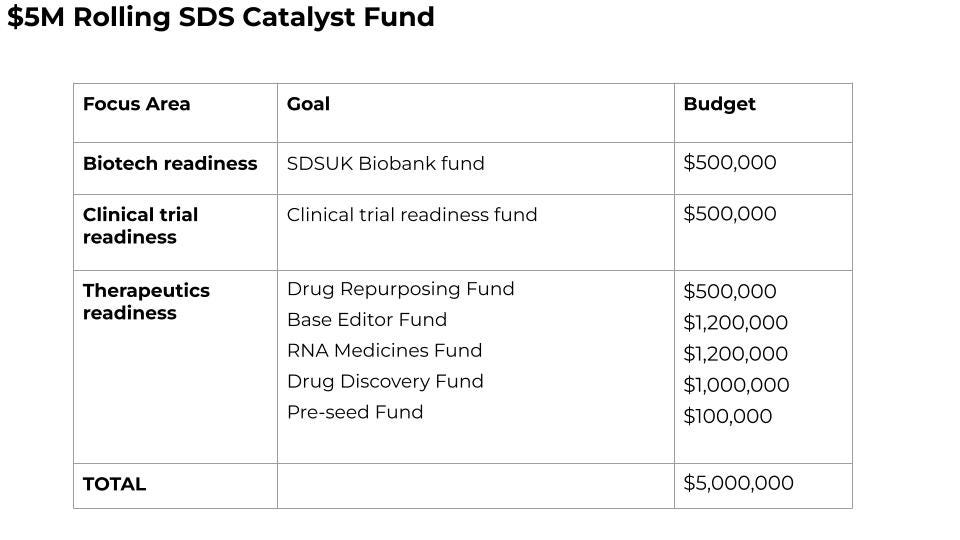
The SDS Cure Roadmap envisions the formation of a SDS Catalyst Fund initially housed within SDSUK and jointly operated with Perlara. At the appropriate time, the SDS Catalyst Fund will be spun out of SDSUK as a separate entity. SDS Catalyst Fund will pool capital and coordinate research projects globally, leverage non-dilutive funding sources, create communal resources, and concurrently de-risk multiple therapeutic tracks to encourage outside investment and biopharma sponsorship. Those activities and capabilities are the keystone species that allow a sustainable SDS-focused biotech ecosystem to develop curative medicines and life-saving prognostic tests.
The umbrella SDS Catalyst Fund will raise and then manage a series of thematic funds. Wherever possible, the Fund will seek matching or supplemental funds from government and foundation grants. For the therapeutics-focused tracks, the Fund will seek sponsorship and in-kind support from interested biotech companies. Individual thematic funds may be constituted as a venture philanthropy fund that makes investments in exchange for equity versus non-dilutive grants with no strings attached.
A $500,000 SDS Clinical Trial Readiness Fund will fund a UK-based definitive longitudinal natural history study using best clinical practices informed by other country-specific foundations.
A $500,000 SDSUK Biobank Fund will fund the creation of a 50-cell panel of fibroblasts, PBMCs and undifferentiated iPSCs from UK-based SDS patients of diverse population backgrounds that will be subjected to multi-omics analyses with the stipulation that all data must be made freely available to any researcher anywhere in the world.
A $500,000 milestone-based SDS Drug Repurposing Fund will be raised to support multi-species and multi-cell type screening projects with the goal of identifying a repurposable clinical candidate for single-patient INDs in under one year.
A $1,200,000 milestone-based SDS Base Editor fund to achieve preclinical proof-of-concept with an investigational 258+2T→C base editor in three of three patient cell models (fibroblasts, PBMCs, iPSCs) and in the humanized SBDS mouse model.
A $1,200,000 milestone-based SDS Nucleic Acids Fund divided 50/50 to achieve preclinical proof-of-concept with an investigational 258+2T→C ASO and an investigational 183-184TA→CT ACE-tRNA therapy in three of three patient cell models (fibroblasts, PBMCs, iPSCs) and in the humanized SBDS mouse model.
A $1,000,000 milestone-based Drug Discovery Fund will be raised to support screening 250,000 novel compound libraries using the top-performing patient-derived cell model and top-performing invertebrate model in the drug repurposing stage.
A $100,000 Pre-Seed Fund, to allow $10,000-$50,000 grants for very early, innovative therapeutic ideas, whether in academic or industry, to ensure the SDS Catalyst Fund does not become tunnel visioned and miss left-field opportunities.
In total, $5M will be required over the next 18 months to achieve clinical proof-of-concept for the drug repurposing track and preclinical proof-of-concept across all the other three tracks. Once projects from any of the four therapeutic tracks achieve preclinical proof-of-concept and foundational IP has been secured, an asset-centric virtual biotech startup accelerator will be pursued to provide seed funding to programs to advance to clinical proof-of-concept.


OPERATING STRUCTURE
This Roadmap envisions that SDSUK will incubate the SDS Catalyst Fund with support from Perlara, and when the time is appropriate, SDSUK will spin out the Fund as an independent and agile legal entity. The SDS Catalyst Fund will raise capital and fund projects across the different work streams listed above. It will be co-managed day-to-day by SDSUK and Perlara. SDS Catalyst Fund will receive equity and/or stock options in biotech companies formed with seed investment, IP or data funded by the Collective. Proceeds from those investments will be re-invested in the long tail and in ensuring patients get access to curative medicines and prognostic tests regardless of where they live or their economic circumstances, in particular when market forces fail to mobilize in spite of commercial successes.
The two founding team members, SDSUK and Perlara PBC, will contribute project management, communications support, and deal assessment totalling 20-30 hours per month at a fair-market nonprofit hourly consulting rate. A wider circle of supporting team members, including reviewers of this Roadmap, will convene virtually every quarter to review research proposals, and eventually fundraising decks and investment memos. Decisions will be made with each reviewer having a vote.
In parallel, SDSUK is working with SDS Foundation and will work with any allied organizations or families. Invitation is open to anyone who would like to participate. Lay versions of all investment committee documents will be circulated in advance of meetings. SDS Catalyst Fund will serve as the coordinating organization between the various SDS foundations and charities around the world.
A federated data platform for consolidating patient registries will be managed by SDS Catalyst Fund with input and guidance from other SDS organizations that wish to participate. The benefit for all SDS registries to collaborate is that all financial returns that are generated by the SDS Catalyst Fund will be funnelled back into SDS research toward the long tail.
RELEVANT EXAMPLES FROM OTHER RARE DISEASES
Patient organization getting economic value for data and reinvesting in mission
CF Foundation – either as part of a venture philanthropy deal with funding and other elements of value being contributed as well or, in the absence of a funding deal, charging a fee for access to registry and other enabling preclinical or patient data.
CF Trust enters into commercial contracts with biopharma to provide the UK patient data registry for Phase 4/pharmacovigilance studies (post-approval obligation to EMA). It saves biopharma building their own database, which is expensive and time consuming, and obviously stops our patient data becoming fragmented.
Foundation-run venture funds
Juvenile Diabetes Research Foundation T1D Fund - $125M raised from donors who get tax benefits + co-investment rights
CureDuchenne Ventures – funded from their share of proceeds from Prosensa’s sale to Biogen after funding exon-skippers
Myeloma Investment Fund – $50M run by Multiple Myeloma Research Foundation with donor money
BEYOND SDS
Models, methods, medicines and tests developed for SDS will have broader applicability. There are the most obvious disease indication expansion opportunities in hematological cancer: myelodysplastic syndrome and leukemia. There are also leukemia predisposition syndromes such as Diamond-Blackfan anemia and Li Fraumeni syndrome. Some mutation-agnostic therapies like an eIF6 inhibitor might have therapeutic benefit in other monogenic ribosomopathies.
The availability of SDS invertebrate models will spur academic researchers to investigate the roles of SBDS and eIF6 in regulating longevity and possibly even healthspan extension. This idea is based on a phenomenon called translational attenuation (Solis et al., 2018).
SDS LONG TAIL
A long tail addendum will be drafted with a focus on helping all SDS patients receive a genetically confirmed and definitive diagnosis and highlighting cure paths for each gene mutation.
ROADMAP UPDATES
This Roadmap is considered version 1 (v1) and it will require annual comprehensive reviews to incorporate progress reports and course corrections.
REFERENCES
Austin KM, Leary RJ, Shimamura A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood. 2005 Aug 15;106(4):1253-8. doi: 10.1182/blood-2005-02-0807. Epub 2005 Apr 28. PMID: 15860664; PMCID: PMC1895203.
Austin KM, Gupta ML Jr, Coats SA, Tulpule A, Mostoslavsky G, Balazs AB, Mulligan RC, Daley G, Pellman D, Shimamura A. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J Clin Invest. 2008 Apr;118(4):1511-8. doi: 10.1172/JCI33764. PMID: 18324336; PMCID: PMC2263145.
Bezzerri V, Bardelli D, Morini J, Vella A, Cesaro S, Sorio C, Biondi A, Danesino C, Farruggia P, Assael BM, D'amico G, Cipolli M. Ataluren-driven restoration of Shwachman-Bodian-Diamond syndrome protein function in Shwachman-Diamond syndrome bone marrow cells. Am J Hematol. 2018 Aug;93(4):527-536. doi: 10.1002/ajh.25025. Epub 2018 Feb 9. PMID: 29285795.
Bezzerri V, Api M, Allegri M, Fabrizzi B, Corey SJ, Cipolli M. Nonsense Suppression Therapy: New Hypothesis for the Treatment of Inherited Bone Marrow Failure Syndromes. Int J Mol Sci. 2020 Jun 30;21(13):4672. doi: 10.3390/ijms21134672. PMID: 32630050; PMCID: PMC7369780.
Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003 Jan;33(1):97-101. doi: 10.1038/ng1062. Epub 2002 Dec 23. PMID: 12496757.
Donadieu J, Leblanc T, Bader Meunier B, Barkaoui M, Fenneteau O, Bertrand Y, Maier-Redelsperger M, Micheau M, Stephan JL, Phillipe N, Bordigoni P, Babin-Boilletot A, Bensaid P, Manel AM, Vilmer E, Thuret I, Blanche S, Gluckman E, Fischer A, Mechinaud F, Joly B, Lamy T, Hermine O, Cassinat B, Bellanne-Chantelot C, Chomienne C, French Severe Chronic Neutropenia Study Group. Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica 2005;90(1):45-53; https://doi.org/10.3324/%25x.
Finch AJ, Hilcenko C, Basse N, Drynan LF, Goyenechea B, Menne TF, González Fernández Á, Simpson P, D’Santos CS, Arends MJ, Donadieu J, Bellanné-Chantelot C, Costanzo M, Boone C, McKenzie AN, Freund SM, Warren AJ. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes and Development (2011) 25: 917-929. PMID: 21536732
Hamabata T, Umeda K, Kouzuki K, Tanaka T, Daifu T, Nodomi S, Saida S, Kato I, Baba S, Hiramatsu H, Osawa M, Niwa A, Saito MK, Kamikubo Y, Adachi S, Hashii Y, Shimada A, Watanabe H, Osafune K, Okita K, Nakahata T, Watanabe K, Takita J, Heike T. Pluripotent stem cell model of Shwachman-Diamond syndrome reveals apoptotic predisposition of hemoangiogenic progenitors. Sci Rep. 2020 Sep 9;10(1):14859. doi: 10.1038/s41598-020-71844-8. Erratum in: Sci Rep. 2021 Jan 18;11(1):2107. PMID: 32908229; PMCID: PMC7481313.
Kanprasoet W, Jensen LT, Sriprach S, Thitiananpakorn K, Rattanapornsompong K, Jensen AN. Deletion of Mitochondrial Porin Alleviates Stress Sensitivity in the Yeast Model of Shwachman-Diamond Syndrome. J Genet Genomics. 2015 Dec 20;42(12):671-84. doi: 10.1016/j.jgg.2015.09.004. Epub 2015 Sep 25. PMID: 26743985.
Kennedy AL, Myers KC, Bowman J, Gibson CJ, Camarda ND, Furutani E, Muscato GM, Klein RH, Ballotti K, Liu S, Harris CE, Galvin A, Malsch M, Dale D, Gansner JM, Nakano TA, Bertuch A, Vlachos A, Lipton JM, Castillo P, Connelly J, Churpek J, Edwards JR, Hijiya N, Ho RH, Hofmann I, Huang JN, Keel S, Lamble A, Lau BW, Norkin M, Stieglitz E, Stock W, Walkovich K, Boettcher S, Brendel C, Fleming MD, Davies SM, Weller EA, Bahl C, Carter SL, Shimamura A, Lindsley RC. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat Commun. 2021 Feb 26;12(1):1334. doi: 10.1038/s41467-021-21588-4. PMID: 33637765; PMCID: PMC7910481.
Lueck JD, Yoon JS, Perales-Puchalt A, Mackey AL, Infield DT, Behlke MA, Pope MR, Weiner DB, Skach WR, McCray PB Jr, Ahern CA. Engineered transfer RNAs for suppression of premature termination codons. Nat Commun. 2019 Feb 18;10(1):822. doi: 10.1038/s41467-019-08329-4. PMID: 30778053; PMCID: PMC6379413.
Menne TF, Goyenechea B, Sánchez-Puig N, Wong CC, Tonkin LM, Ancliff PJ, Brost RL, Costanzo M, Boone C, Warren AJ. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007 Apr;39(4):486-95. doi: 10.1038/ng1994. Epub 2007 Mar 11. PMID: 17353896.
Oyarbide U, Shah AN, Amaya-Mejia W, Snyderman M, Kell MJ, Allende DS, Calo E, Topczewski J, Corey SJ. Loss of Sbds in zebrafish leads to neutropenia and pancreas and liver atrophy. JCI Insight. 2020 Sep 3;5(17):e134309. doi: 10.1172/jci.insight.134309. PMID: 32759502; PMCID: PMC7526460.
Elayne Provost,1 Karen A. Wehner,3,* Xiangang Zhong,1,* Foram Ashar,2 Elizabeth Nguyen,4 Rachel Green,3 Michael J. Parsons,2 and Steven D. Leach1,2,‡Ribosomal biogenesis genes play an essential and p53-independent role in zebrafish pancreas development. Development. 2012 Sep 1; 139(17): 3232–3241. doi: 10.1242/dev.077107. PMCID: PMC3413166, PMID: 22872088
Solis GM, Kardakaris R, Valentine ER, Bar-Peled L, Chen AL, Blewett MM, McCormick MA, Williamson JR, Kennedy B, Cravatt BF, Petrascheck M. Translation attenuation by minocycline enhances longevity and proteostasis in old post-stress-responsive organisms. Elife. 2018 Nov 27;7:e40314. doi: 10.7554/eLife.40314. PMID: 30479271; PMCID: PMC6257811.
Takai A, Chiyonobu T, Ueoka I, Tanaka R, Tozawa T, Yoshida H, Morimoto M, Hosoi H, Yamaguchi M. A novel Drosophila model for neurodevelopmental disorders associated with Shwachman-Diamond syndrome. Neurosci Lett. 2020 Nov 20;739:135449. doi: 10.1016/j.neulet.2020.135449. Epub 2020 Oct 25. PMID: 33115644.
Tan S, Kermasson L, Hoslin A, Jaako P, Faille A, Acevedo-Arenzana A, Lengline E, Ranta D, Poirée, M, Fenneteau O, Ducou le Pointe H, Fumagalli S, Beaupain B, Nitschke P, Bôle-Feysot C, de Villartay J-P, Bellanné-Chantelot C, Donadieu J, Kannengiesser C, Warren AJ*, and Revy P*. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood 2019 Jul 18;134(3):277-290. doi: 10.1182/blood.2018893404. Epub 2019 May 31 PMID: 31151987
Shengjiang Tan,1,2,3,*, Laëtitia Kermasson4,*, Christine Hilcenko1,2,3, Vasileios Kargas1,2,3, David Traynor1,2,3, Ahmed Z Boukerrou1,2,3, Norberto Escudero-Urquijo1,2,3, Alexandre Faille1,2,3, Alexis Bertrand4, Maxim Rossmann1,2,3, Beatriz Goyenechea3,§, Li Jin3,ø , Jonathan Moreil4, Olivier Alibeu5, Blandine Beaupain6, Christine Bôle-Feysot5, Stefano Fumagalli7,8, Sophie Kaltenbach9,10, Jean-Alain Martignoles11, Cécile Masson12, Patrick Nitschké12, Mélanie Parisot5, Aurore Pouliet5, Isabelle Radford-Weiss9,10, Frédéric Tores12, Jean-Pierre de Villartay4, Mohammed Zarhrate5, Ai Ling Koh13,14, Kong Boo Phua13,14, Saumya S Jamuar13,14, Peter J Bond15,16, Christine Bellanné-Chantelot17, Isabelle Callebaut18,°, François Delhommeau11,°, Jean Donadieu19,°, Alan J Warren1,2,3,@,#, Patrick Revy4,@,#. Somatic genetic rescue of a germline ribosome assembly defect. Nature Communications (2021) 12, 5044. https://doi.org/10.1038/s41467-021-24999-5
Tulpule A, Kelley JM, Lensch MW, McPherson J, Park IH, Hartung O, Nakamura T, Schlaeger TM, Shimamura A, Daley GQ. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell. 2013 Jun 6;12(6):727-36. doi: 10.1016/j.stem.2013.04.002. Epub 2013 Apr 18. PMID: 23602541; PMCID: PMC3755012.
Venkatasubramani N, Mayer AN. A zebrafish model for the Shwachman-Diamond syndrome (SDS). Pediatr Res. 2008 Apr;63(4):348-52. doi: 10.1203/PDR.0b013e3181659736. PMID: 18356737.
Wong CC, Traynor D, Basse N, Kay RR, Warren AJ. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood. 2011 Oct 20;118(16):4305-12. doi: 10.1182/blood-2011-06-353938. Epub 2011 Jul 29. PMID: 21803848.
Weis F, Giudice E, Churcher M, Jin L, Hilcenko C, Wong CC, Traynor D, Kay RR, Warren AJ. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nature Struct Mol Biol. 2015 Nov;22(11):914-9. doi: 10.1038/nsmb.3112. Epub 2015 Oct 19. PMID: 26479198; PMCID: PMC4871238.
Zhang S, Shi M, Hui CC, Rommens JM. Loss of the mouse ortholog of the shwachman-diamond syndrome gene (Sbds) results in early embryonic lethality. Mol Cell Biol. 2006 Sep;26(17):6656-63. doi: 10.1128/MCB.00091-06. PMID: 16914746; PMCID: PMC1592835.