Leigh Syndrome drug repurposing updates
In collaboration with Cure Mito Foundation and using a SURF1-deficient yeast model, we're validating hits from the pilot screen, and we just screened a quarter of the comprehensive ReFRAME library.


In collaboration with



We’ve been working with Cure Mito Foundation on a yeast-powered drug repurposing/multipurposing project because of the extraordinary evolutionary conservation of the electron transport chain in mitochondria across all animals, including the single-cell fungus we affectionately call baker’s yeast aka Saccharomyces cerevisiae. Although we are by no means the first scientists to be enthusiastic about the potential of “simple” yeast models of Leigh Syndrome, we seem to be among only a handful who are acting on this conviction. Why?
Good question!
We’ve known for a long time that the mitochondria that reside inside yeast cells behave like their sisters inside human cells. For example, when yeast are grown on a non-fermentable carbon source like glycerol, they are forced to respire, i.e., put their mitochondria to work. In response to glycerol fuel, the mitochondria mushroom into a cell-filling tubular network, expanding the total mitochondrial membrane surface area and volume by 300% in order to enable all the electron transport that terminates at Complex IV aka cytochrome c oxidase (COX).
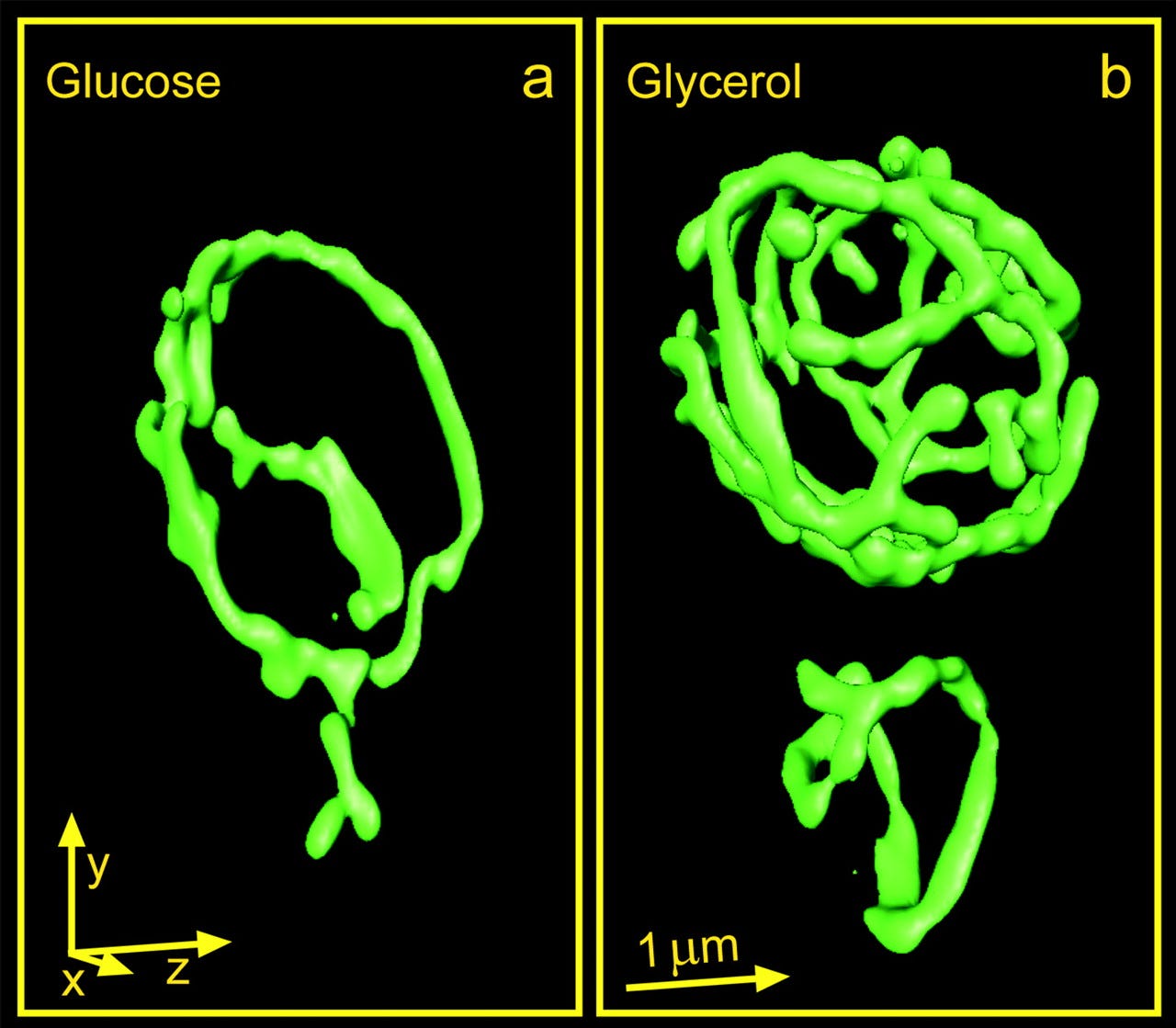
The COX active site is rendered in the featured image: the reactor core where molecular oxygen is reduced to water with the assistance of precisely positioned copper and iron ions. The SURF1 nuclear gene encodes a mitochondrial inner membrane protein required for COX assembly and activity.
An intuitive analogy is an expert yogi who helps you assume the correct posture — in protein-speak it’s called a “conformation” — down to the exact angles of your joints. Without SURF1, the inherently wobbly COX active site is not stabilized in 3-D space and so cannot efficiently allow the final electron handoff to oxygen to occur.
We set out to identify repurposable compounds that could compensate for Complex IV deficiency caused by complete loss of SURF1. Compensation could take the form of a SURF1 bypass pathway. For example, boosting the function another part (protein subunit) of Complex IV. A more extreme example of a SURF1 bypass pathway would be somehow rerouting the flow of electrons to oxygen, seemingly a tall order.
Alternatively, compensation by the paradoxical “two wrongs make a right” mechanism would involve slowing down upstream electron transport by Complexes I to III to match the hobbled catalytic capacity of Complex IV. We’ve seen this type of metabolic flux rebalancing in a yeast model of PMM2-CDG, one of the congenital disorders of glycosylation (CDGs) that we work on.
There’s a third possibility. Compensation could take the form of small molecule chaperones that bind directly to COX. Pharmacological chaperones are a well-known class of small molecule drug. For example, Kalydeco is the first pharmacological chaperone approved for cystic fibrosis. It boosts the function of the mutant CFTR protein. In other words, there must be a mutant protein present in the cell that can be stabilized by a traditional small molecule chaperone.
However, most SURF1 pathogenic loss-of-function mutations result in a truncated or absent protein. The most common recurring SURF1 mutation, which mirrors the effect of SURF1 whole-gene deletion, is c.312_320del10insAT, resulting in Leu105X. Therefore in a SURF1-knockout yeast, there is no residual SURF1 protein for a small molecule chaperone to act on. In theory, a small molecule chaperone could act as pharmacological prostheses that stabilizes the COX active site. You’ve heard of gene replacement therapy: this would be pharmacological replacement therapy.
The whole point of an unbiased screen is to let nature tell us what’s possible rather than making what we think is a really educated guess about what should or shouldn’t work as a therapeutic mechanism.
We completed the 2,250-compound Pharmakon library screen a few months ago, as previously summarized here.
Let’s recap the data we’ve collected thus far. Recall that we screened a haploid SURF1-deficient mutant and a diploid SURF1-deficient mutant in parallel to ensure replicability and robust assay performance. We also wanted to make sure there weren’t any unexpected discrepancies attributable to ploidy, i.e., yeast cells expressing one copy of every chromosome versus yeast cells expressing two copies of every chromosome.
Here’s the Z-score distributions for both haploid and diploid shy1∆ (SURF1-knockout) mutants.
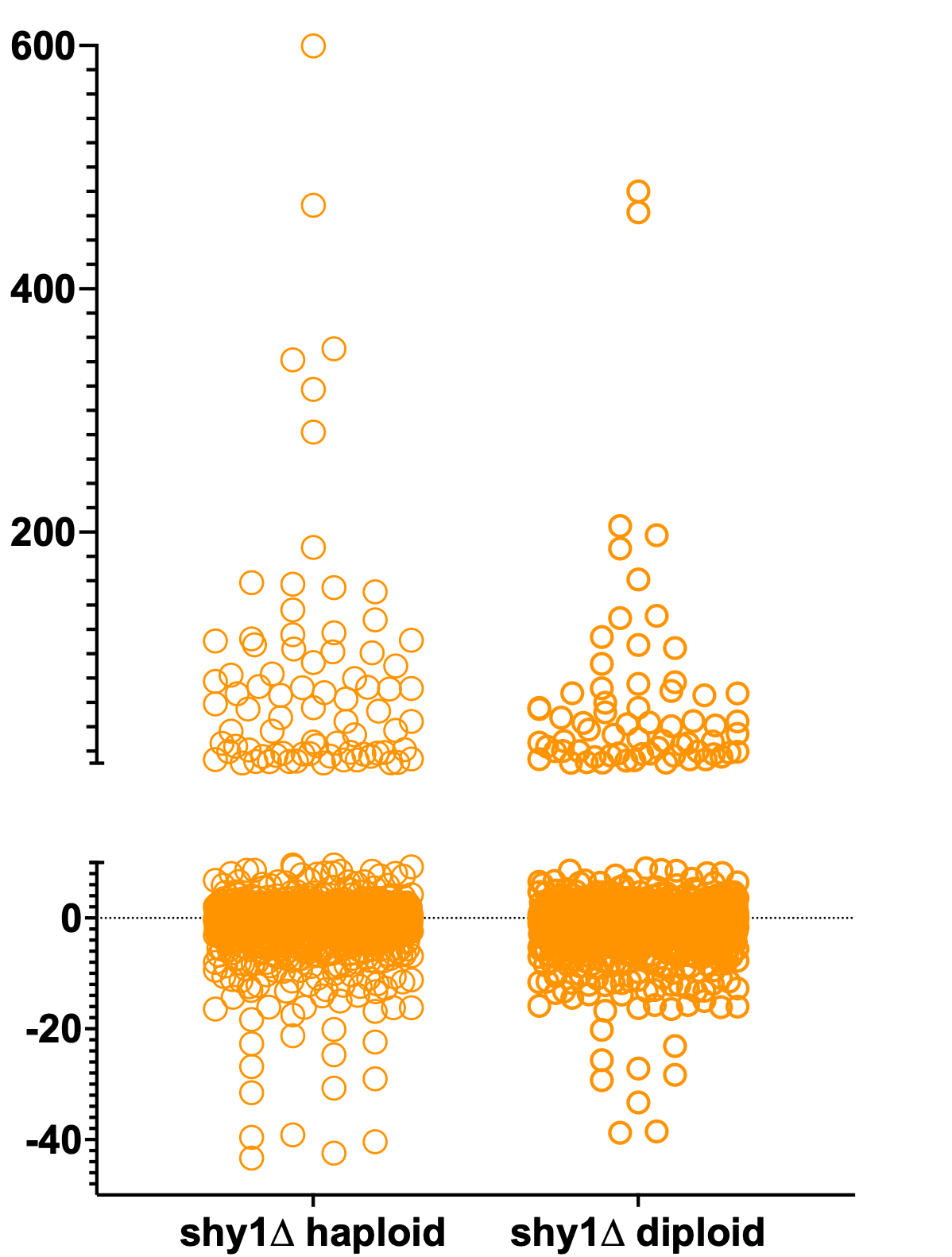
As another quality control step, we plotted the haploid data versus the diploid data. There is only a single test compound that is a hit for the haploid knockout mutant but not for the diploid knockout mutant. Otherwise the correlation is tight.

We’re not quite ready to share the compound IDs and structures yet since hit validation studies to assess whether the rescuers boost cytochrome c oxidase activity in yeast cells are still pending. We can share the following teaser though.
Professor Alessandro Prigione’s lab, whose lab is also funded by Cure Mito Foundation, conducted an independent drug repurposing screen using Leigh Syndrome patient derived iPSC neurons (iNeurons). Their top hit belongs to the latest generation of a storied drug class whose antecedents are rescuers of SURF1-deficient yeast. Thanks to Cure Mito, we’re pooling our data and working on a combined paper.
We saw this type of convergence between a yeast model and an iAstrocyte model of PGAP3. By our estimates, the cost per compound of screening a yeast model is currently around $1. The cost per compound of screening an iPSC-derived differentiated cell model is 50-350X more expensive. Ideally the yeast model is the high-throughput battering ram that narrows thousands of compounds to dozens. Those dozens of hits can then be tested in costlier and more finnicky iPSC models for low-throughout mechanistic validation studies.
Last week we completed the first quarter of the ReFRAME library — another thirty-six 384-well plates to go. As expected, we see complete separation between the negative control wells and the positive control wells. We also observed rescuers/hits!
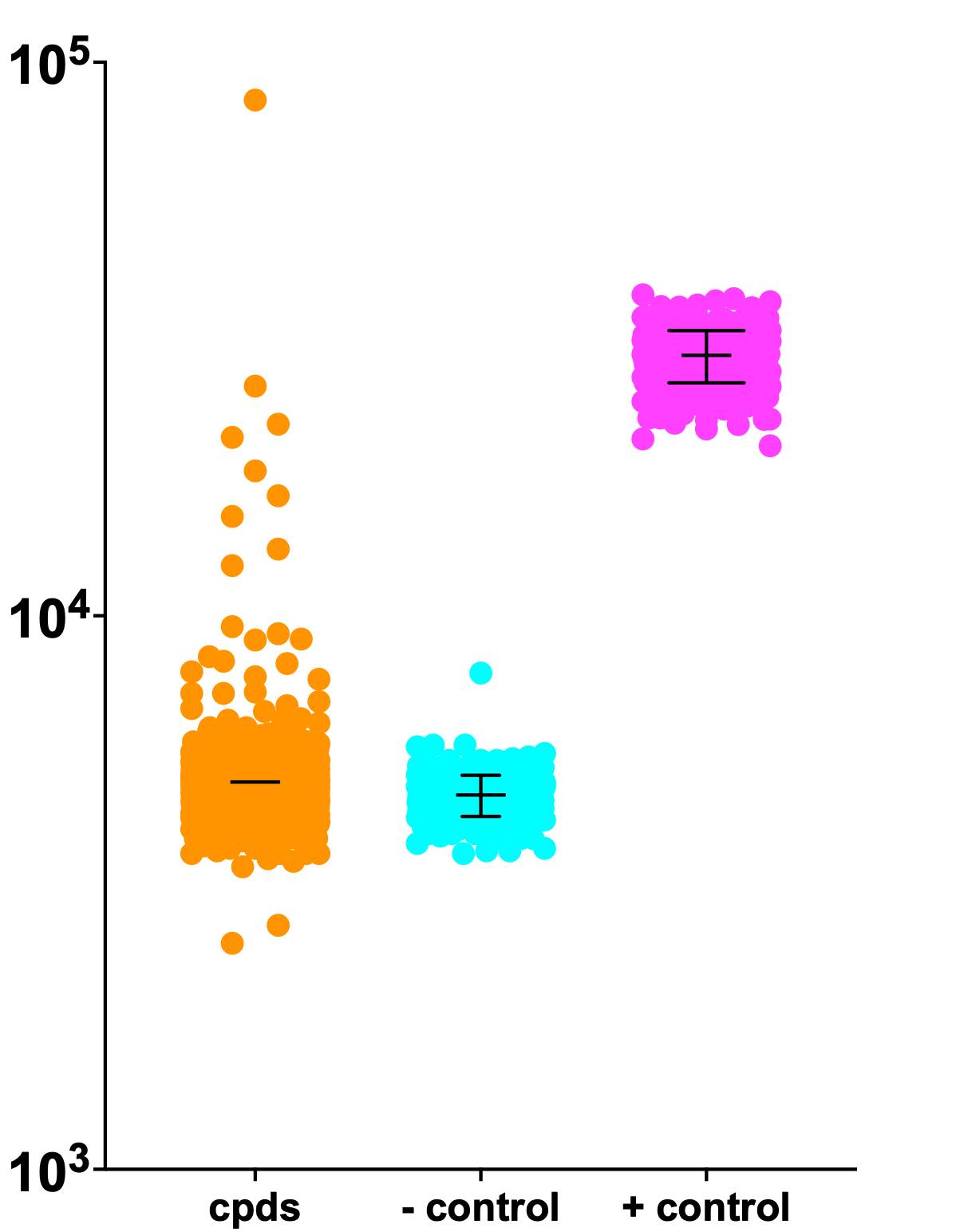
We’ll complete the remainder of the ReFRAME library in the next 1-2 weeks. In parallel, we’ll complete the Pharmakon hit validation biochemical studies, which involves physically isolating mitochondria from drug-treated SURF1-deficient yeast cells and then assaying COX enzymatic activity.
Finally, we’re excited to share that we’ll be screening six more Leigh Syndrome/LS genes as we expand our collaboration with Cure Mito Foundation beyond just SURF1. The next LS gene currently in the queue is AFG3L2, for which initial growth characterization and seeding density experiments have been completed.
More to come soon!