NARS1 Cure Odyssey
A little girl who didn’t make it to her second birthday provided the inspiration for a foundation that is trying to make life better for those who share her genetic disorder.



In collaboration with



 / Twitter")
Rory Belle started showing symptoms very early in life, which concerned her parents. And her mother, Rachel Heilmann, whose background was in clinical pharmacology, knew what she wanted for her child. But knowing and getting are two different things, and Rory Belle lost her life to a genetic variant in the gene NARS1, barely nine months after her diagnosis.
Sitting in a hospital room on the 4th of July in 2020, her liver enzymes were 700, you could see her liver. A geneticist came in with the results of exome sequencing. We didn’t know anything about it. We would google NARS1 and there was nothing out there.
Rachel Heilmann
Like so many parents dealing with rare diseases, the internet was the first place Rachel went, which eventually brought Rachel in contact with others dealing with the same issue — in this case, another mother, Teri Declercq, whose son Grady was diagnosed when he was three years old, following many rounds of testing and re-testing. After months of reaching out to researchers who looked to have some connection with NARS1, Rachel finally heard back from a group at University College London, Henry Houldlen and his post-doc Stephanie Efthymiou, who had collected data on around 10% of NARS1 cases. After Stephanie and Rachel spoke, they decided to start a FaceBook page. “That’s where our story begins,” says Rachel.
What is NARS1?
The NARS1 gene encodes the asparagine aminoacyl tRNA synthetase gene, one of twenty such enzymes that operate in the cytoplasm to charge tRNAs with their cognate amino acids. This being an essential step in protein synthesis, these enzymes (ARSs) are found ubiquitously and highly conserved in nature. Mutations in a few of them have been reported to lead to neurological problems — developmental and intellectual delays, movement difficulties, seizures, being the most common — which vary in severity. Notably, NARS1 stands out among the ARSs with its neurodevelopmental delay and multi-system presentation.
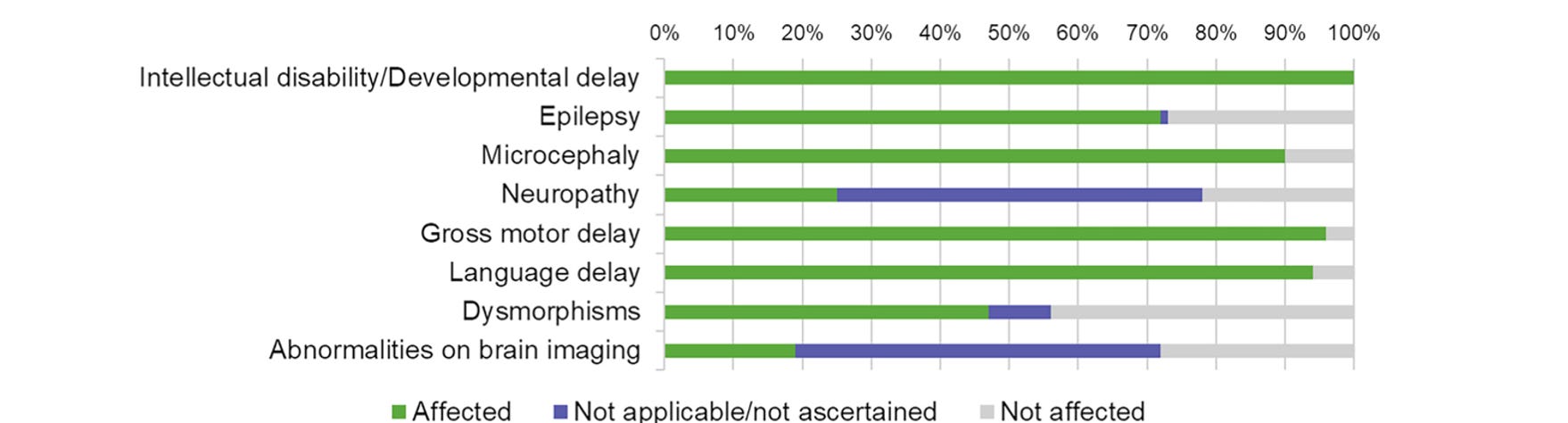
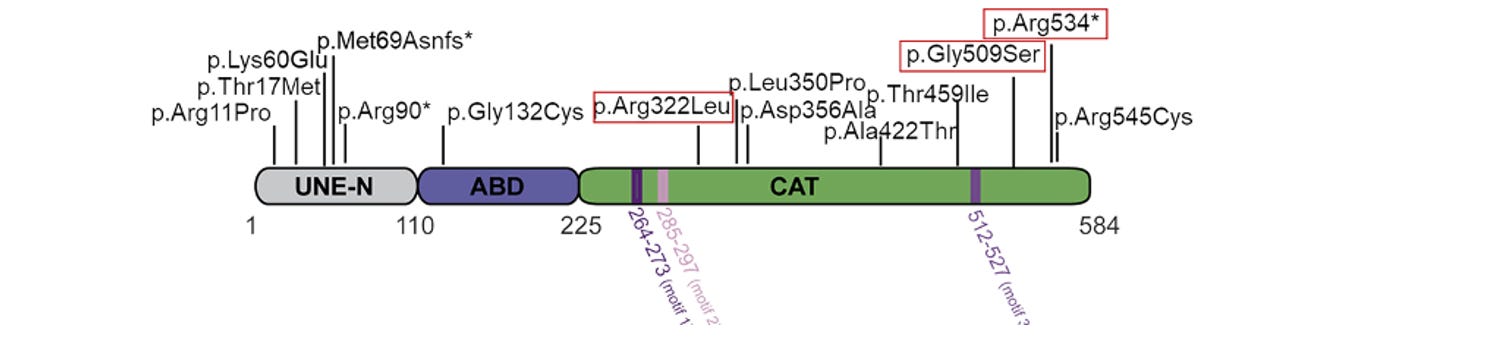
The NARS1 gene codes for a 548 amino acid protein, located in humans on chromosome 18. It has three domains: a catalytic domain (CAT), which charges the tRNA with its cognate amino acid, an anticodon binding domain (ABD) which positions the tRNA at its appropriate place along the mRNA, and a unique domain (UNE-N). At least, two forms of NARS1 disorders exist, toxic gain of function variants which are believed to be de novo mutations that cause progressive peripheral neuropathy, and recessive partial loss of function mutations, which are in most cases heterozygote (bi-allelic) mutations that affect brain development in particular.
What researchers have discovered about the mutations so far are that a number map to two mutational hotspots (arginine 534 and arginine 545) located within the last 40 amino acids of the protein. Mutations close to the beginning of the protein lead to severe phenotypes, and other mutations are scattered throughout the gene, as shown below:
Where NARS1 disorder differs from other tRNA synthetase mutations is that there is a clear CNS phenotype including severe neurodevelopmental delay in some cases, but also a peripheral neuropathy, which has shown up in all the kids so far, according to Rachel. This puts NARS1 in a similar camp as Charcot-Marie Tooth disease, a common neurological disease that causes peripheral neuropathy.
“We have that and plus all the things happening in the brain and how it is functioning, and communicating with the rest of the body like walking, talking, eating, GI issues. We’re starting to peel back some of those layers and make connections with other researchers studying our gene and have invested time in other ARS genes."
Rachel Heillman
One life forever changed and another never to be forgotten
Rory Belle’s life and death have changed the course of her mother’s life, even the way she talks about her life. Rachel’s marks time by how long after Rory Belle died some event occurred. Not content with the work she had been doing before Rory Belle came into this world — work that had no connection to her life and her loss — Rachel quit her job in pharma and eventually found herself working with several non-profits, all of which are tackling rare diseases in different combinations. And in 2022, Rachel and Teri founded the Rory Belle Foundation.
In the beginning, most of the positions were held by family members, but the group has grown and diversified, as more families with different backgrounds and expertise become part of the NARS1 family. In the year and a half since its foundation, they have been figuring out the lay of the land — who you need to talk to, where you need to go, when you get money, what you need to do with it, the order of operations. Just trying to sort it all out.
We’re still small but we’re large enough and gotten enough of the right people involved with the right skills, researchers, clinicians, and families have stepped up to help. In service of the kids—to help every kid.
Rachel Heillman
Each ultra-rare disease family is like a firefly. Fragile and floating through the forest, solitary and in search of solidarity. Invisible to everyone else except fellow firefly families, off in the distance. One teams up with a friend to make two. More and more come together. A speck becomes a spark. Ten thousand sparks shine like a sun.

The metaphor extends beyond families to communities. Ultra-rare disease groups are also fireflies self-organizing in the night quietly, unrelentingly, undeterred by obstacles or darkness. Then the light ignites, illuminating corridors that lead to treatments and cures.
Drug repurposing is the first door that needs to be opened. Over the past two years, Perlara working alongside pioneer families has amassed laboratory and real-world data proving that yeast avatars are therapeutically relevant and predictive disease models for patients with inherited metabolic diseases.
On paper, NARS1 is a near 10/10 fit for disease modeling and drug repurposing using yeast avatars. NARS1 is required for an essential metabolic process that operates at the molecular level nearly identically in cells of the human body and in yeast cells: the assembly of proteins, stitched together one amino acid at a time. If proteins were constructed out of Legos, NARS1 would be one of 20 essential bricks.
Not surprisingly, deleting DED81, the yeast version of human NARS1 gene, is lethal to yeast. The killer experiment, as they say in the lab, demonstrates that the human NARS1 gene complements (can replace) the yeast NARS1 gene while NARS1 constructs containing known pathogenic variants do not complement.
In other words, yeast cells cannot grow if the DED81 gene is deleted, but giving back a healthy copy of the human NARS1 gene restores the ability of DED81-deficient yeast cells to grow. The effect requires a functional NARS1 protein because non-functional copies of NARS1 fail to rescue growth.

We kindly received the ded81-5001 temperature-sensitive mutant yeast strain from the Boone lab. As expected, initial growth characterization experiments showed that the ded81-5001 mutant exhibits exquisite heat sensitivity at 38˚C. In other words, the ded81-5001 mutant is unable to grow at high temperature. Our drug repurposing goal was to find compounds that, like Lazarus, would bring yeast cells on the brink of death back to life.
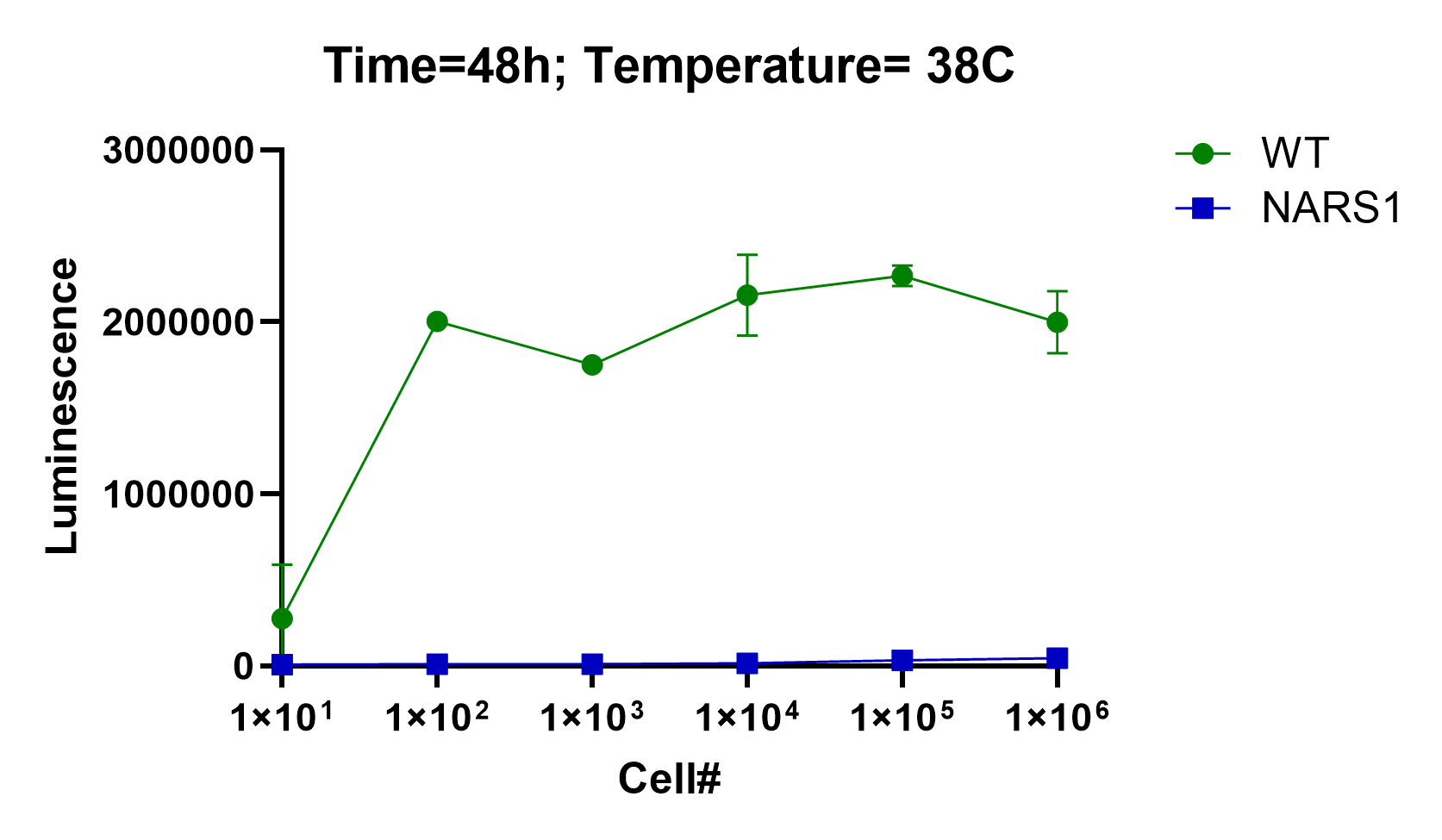
Next on the yeast drug repurposing workflow checklist: seeding density experiments. The initial growth characterization experiment is a quick-and-dirty spot check that ensures the mutant strain is sick. We always test multiple incubation timepoints, e.gs., 24 hours and 48 hours. We also test several temperatures when working with heat-sensitive mutants. Sometimes we’ve gotten tripped up at this step. In the case of NARS1, the ded81-5001 temperature-sensitive mutant yeast strain performed to expectations.
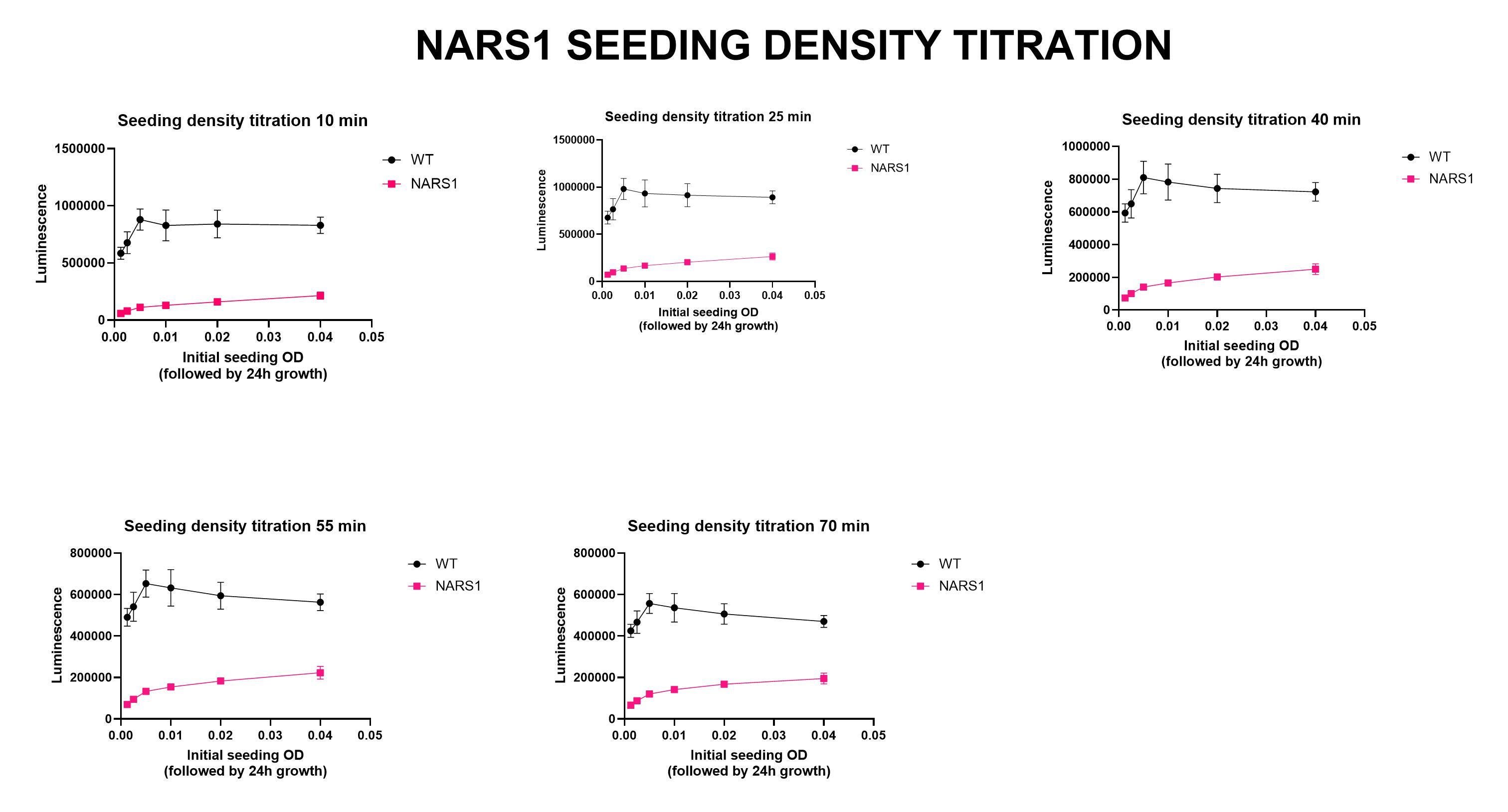
The seeding density experiments locks in the most critical variables of yeast cell density and the incubation time for the lumiscence-based growth assay. The final pre-screening step is the Z’ optimization experiment. Our target Z’ value is any number greater than 0.5, which in this case we achieved at the lowest concentration of yeast cells (OD 0.0025) and after the shortest incubation time post-cell lysis (10 minutes).

All checks were a go — so we progressed to the TargetMol screen.
Below is the Z-score distribution of the 8,400-compound TargetMol library screen.
Each of the TargeMol compounds is an orange point. The gray points in the upper segment of the y-axis are the ATP and ADP positive controls, or benchmarks that establish the assay ceiling. The four gray points in the lower segment of the y-axis are artifacts of one of the ATP wells that creates a cross-shaped halo.

The cyan points (negative control) are the ded81-5001 temperature-sensitive mutant yeast strain treated with DMSO, which can be considered a placebo. The negative control wells all performed as expected. The magenta points (positive control) are the wildtype yeast strain. The positive controls on one of the assay plates underperformed. Those points can be disregarded.
Reminiscent of PIGN, PIGA, ALG11, ADSL and DHDDS, the rescue ceiling is low. Meaning the rescuers allow the heat-shocked and extremely sick ded81-5001 mutant strain to grow to numbers that are several fold greater than the negative control (placebo-treated) ded81-5001 mutant strain, but that growth benefit translates to only a small percentage of the total growth achieved by the wildtype strain.
By contrast, the screens we conducted on SURF1, FARS2, AFG3L2, MT-ATP6, PIGW, PGAP3 resulted in rescuers that rescued mutant strain growth to levels approximating, equating, or even exceeding maximal wildtype growth. Why are mutants that are equally sick more or less rescuable? It’s not clear to us today what factors determine the altitude of the rescue ceiling. But we can ask a simpler question: how does NARS1 compare to other inherited metabolic diseases in yeast?
The obvious first comparison is between NARS1 and FARS2, a fellow ARS gene. NARS1 is an asparagine (abbreviated N) tRNA synthetase, and FARS2 is a phenylalanine (abbreviated F) tRNA synthetase. Two different Lego bricks. Adding another wrinkle of complexity, the mitochondria has its own posse of ARS genes, which usually get the number 2 designation, e.g., FARS2.

Even though NARS1 encodes a cytoplasmic tRNA synthethase and FARS2 encodes a mitochondrial tRNA synthetase, we naively could have assumed that the two ARS diseases share more in common than not. Much to our surprise, NARS1 and FARS2 only share a few rescuers — ATP and ADP are positive controls and are expected to set the assay ceiling. Strikingly, the compounds boxed in orange above rescue FARS2 mutant yeast but make the NARS1 mutant yeast more sick! This was an important clue.
Across the set of 16 different yeast screens completed to date, we’ve noticed a peculiar and provocative pattern. Compounds that rescue mitochondrial mutants make congenital disorder of glycosylation (CDG) mutants more sick. Curiously, we don’t observe the converse, i.e., compounds that rescue CDG mutants and make mitochondrial mutants more sick. That got us thinking how NARS1 would stack up against a CDG.
Surprise number two! NARS1 and ALG11-CDG have a lot in common. The magenta box contains shared rescuers and the cyan box contains shared sensitizers. Those boxes are almost empty in the NARS1 vs FARS2 pairwise comparison. The sensitizers in the cyan box are sensitizers in almost every other TargetMol screen of a CDG mutant.

So little is known about NARS1 that nothing should come as a surprise. Yet how do we connect the dots between NARS1 and congenital disorders of glycosylation? Maybe the answer is staring us in the face. Recall that the N in NARS1 stands for asparagine.
Guess where most of the sugars that decorate proteins are attached? The N in N-linked glycosylation, the most common type of protein glycosylation, also stands for asparagine. What if NARS1 is actually more accurately described pathophysiologically as a type of Congenital Disorder of Glycosylation rather than a type of Charcot-Marie Tooth?
Where there’s smoke, there’s fire. NARS1 and most CDGs feature infant-to-child onset of neurodevelopmental delay and intellectual disability with variable seizures and behavioral disturbances, followed by progressive peripheral neuropathy as kiddos age. Remember that NARS1 stands out among the ARSs clan. It’s the first (and most diagnosed) ARS that primarily presents early in life with multi-organ and neurodevelopmental disorder instead of a later onsetting progressive peripheral neuropathy.
A molecular explanation neatly presents itself. NARS1 deficiency would lead a decrease in the incorporation rate of asparagine residues into nascent proteins, meaning newly minted proteins would have fewer attachment points for glycans (sugar chains). Such a model makes a clear prediction: proteins in a NARS1-deficient cells are under-glycosylated, or hypoglycosylated to use the technical term. A concomitant testable prediction is that asparagine levels will be altered in NARS1 patients.
We presented this results to Rachel and Teri from the Rory Belle Foundation this week and we’re discussing how to proceed with hit validation. Excitingly, several of the top rescuers are clinically actionable nutraceuticals/supplements. Onward for NARS1!