NFIX Cure Roadmap
The 6th Cure Roadmap was commissioned by the Malan Syndrome Foundation. We made the case for drug repurposing and gene up-regulating ASOs, and sparked a collaboration to derive iPSCs from urine.


NFIX-related Malan Syndrome Cure Roadmap
Prepared for Malan Syndrome Foundation by Perlara PBC
Q2 2022

VISION
Towards a future where people with Malan syndrome will lead healthy, interactive, and fulfilled lives. This multi-year, multi-modality roadmap describes the scientific and commercial strategy for finding a medicine for Malan syndrome patients. Based on the current disease understanding of NFIX, we recommend moving forward with two therapeutic approaches for this condition. Furthermore, this Roadmap includes recommendations regarding the Malan Syndrome Foundation’s research portfolio and makes recommendations for prioritization based on the NFIX knowledgebase and the active projects supported by the Malan Syndrome Foundation.
This Roadmap envisions the following milestones and timelines:
· In 6-9 months, the preliminary experimentation will be performed to assess the likelihood for success of the two proposed therapeutic modalities: Drug repurposing and antisense oligo (ASO)-based modalities. This includes testing the activity of an NFIX-responsive gene reporter in a high-throughput drug screen. In parallel, we suggest an in vivo drug-repurposing effort using the worm nfi-1/NFIX model developed by InVivo Biosystems. Preliminary experiments suggest a motion phenotype that might be amenable to high-throughput screening efforts in vivo. As a second track, we advise further testing of the efficiency of various ASO strategies in targeting the NFIX 5’UTR for their capacity to boost NFIX expression levels. Feasibility studies will inform which therapeutic modality may offer the best chance for long-term success.
· In 9-15 months, we will optimize assays for both strategies and provide validation in clinically relevant cell types such as muscle and neurons. Drug screening of NFIX target gene reporters will be underway. If one of the two modalities fails as a therapeutic avenue in the first 6-9 months, we will pivot to developing gene therapy-based precision medicine strategy for replacing the affected Exon 2 of NFIX in Malan patients.
· In 16-24 months, we hope to generate preclinical data packages based on our strategies that will form the basis for tackling fundraising and partnership opportunities with biotech and pharmaceutical companies. As Prof. Messina’s work on muscle NFIX has shown, lowering NFIX levels also has therapeutic potential for addressing a multitude of muscular degeneration diseases such as Duchenne Muscular Dystrophy (DMD). Our Drug Repurposing and ASO strategies will be designed in such a way that we will also pick up candidates that reduce as well as increase NFIX levels. This approach will hopefully incentivize investment from this much larger disease space. Furthermore, we envision expanding existing ties with the CACNA1A and MSS Foundation in order to generate critical mass for fundraising and preclinical data generation efforts.

Authors
Victoria M. Blake, PhD (Cure Guide, Perlara PBC)
Jerome P. Korzelius, PhD (Ass. Prof. at University of Kent, Cure Guide, Perlara PBC)
Ethan O. Perlstein, PhD (CEO of Perlara PBC & Maggie’s Pearl LLC)
Reviewers
We invite post-publication review of this Cure Roadmap on Substack
Executive Summary
The goal of the Malan Syndrome Foundation Cure Roadmap is to stimulate development of medicines that will treat and one day (in combination) cure Malan Syndrome. Furthermore, our Cure Roadmap makes recommendations for the Malan Syndrome Foundation’s research funding portfolio and which projects and features to prioritize to best serve the needs of the community. Perlara’s agile Cure Guide operating structure and a Slack-first, globally distributed team will enable research capacity building and preclinical proof-of-concept studies across multiple programs that will ultimately coalesce into developing new curative medicines. The Malan Syndrome Foundation has generated a very active and collaborative research community that has helped to bring the NFIX knowledgebase to a point that it is now possible to start efforts for the development of therapeutics for the treatment of this condition in the coming 1-2 years. For this Roadmap, the authors have explored the NFIX disease space and collaborations and research projects sponsored by the Malan Syndrome Foundation.
Our key recommendations for the coming 12-24 months for the Malan Syndrome Foundation are:
Therapeutic track 1: Drug repurposing (in vitro): focus on in vitro drug-repurposing screen using a NFIX reporter gene in collaboration with Dr. Kooblall (Oxford University) and MSS Foundation.
Therapeutic track 1: Drug repurposing (in vivo): Use of C. elegans nfi-1/NFIX mutant phenotype as readout for whole-animal drug repurposing screen in collaboration with InVivo Biosystems and/or other partners.
Therapeutic Track 2: ASO precision medicine: Expand funding of Dr. McIntosh’s research to fast-track the design and screening for NFIX-specific ASOs with the goal of upregulating NFIX expression.
Cellular models: Investing in efforts to generate patient-specific iPS cell lines using urine-derived cells in collaboration with UriCell and CombinedBrain to fix shortage of patient-derived cell lines.
Collaboration management: establish active collaborations with MSS and CACNA1A Foundations to pool resources for NFIX drug-repurposing screens.
Collaboration management: Continue community-building efforts to establish an Overgrowth Alliance in collaboration with Prof. Tatton Brown to broaden the knowledgebase around this group of syndromes and to possibly pool resources and fundraising efforts.
The Roadmap below will outline the key considerations behind these recommendations.

Introduction
Malan syndrome is caused by haploinsufficiency of the NFIX gene. NFIX is a transcription factor of the NFI family of transcription factors that regulate cell identity, proliferation and cell death in a variety of tissues, with prominent roles in the CNS, bone, hematopoietic system and muscle. Clinical features of Malan syndrome include CNS-related phenotypes such as macrocephaly, moderate to severe ID, anxiety and several autism-like phenotypes such as hyperacusis (sensitive hearing) and poor sociability. Other features include low muscle tone, coordination difficulties and a variety of vision-related problems such as strabismus (crossed eyes) as well as cortical vision problems. Incidence of Malan syndrome has been estimated to be 1:38,000 (1) and as of December 2021, 143 families are found in the Malan Syndrome Foundation registry.

The NFIX transcription unit encompasses 11 exons and encodes 6 isoforms that differ by alternative splicing of exon 7 and 9 and the use of alternative transcription initiation sites. Malan patient mutations predominantly cluster in exon 2, coding for the DNA-binding domain that is highly conserved between NFI-family members (2). The NFIX gene is also affected in a closely-related, more severe condition called Marshall-Smith Syndrome (MSS). Whereas Malan syndrome mutations mostly cluster in Exon 2, MSS syndrome mutations cluster throughout exons 4-11 in the less-conserved CFI/NFI domain involved in transcriptional activation or repression. Furthermore, Malan syndrome mutations can genetically be classified as haploinsufficiency (50% loss of function) for NFIX, whereas MSS mutations are thought to act in a dominant-negative fashion. Of interest, recent additions to the Malan Syndrome Foundation and the patient cohort of Dr. Manuela Priolo, one of the leading clinicians on Malan Syndrome, has identified the adjacent CACNA1A gene to be affected in several Malan syndrome patients that carry Chr. 19p microdeletions and duplications.
NFIX exerts its function in the cell as a transcription factor (TF) that regulates the expression of genes in specific cell types. Research from NFIX expert researchers such as Profs Gronostajski, Piper, Priolo and Messina has demonstrated the diverse roles of NFIX in different tissues such as the brain, muscle, eye and heart (3). As the effects of NFIX loss of heterozygosity are manifested in many of these diverse tissues, the overall strategy for treatment of Malan Syndrome should focus on the restoration of NFIX levels and/or activity in all tissues of the body. To achieve this, we have prioritized options for drug discovery and will outline our strategic recommendations below.
Diversified portfolio of therapeutic modalities
The inability to predict the success of any one treatment modality necessitates a diversified portfolio of mutation-agnostic and mutation-targeted strategies that are embodied in distinct but combinable therapeutic modalities.
We are fortunate in that the work done by many of the NFIX experts funded by the Malan Syndrome Foundation has generated a wealth of knowledge and tools for the study of NFIX (3). With these tools in hand, the Malan Syndrome Foundation Cure Roadmap envisions advancing toward new medicines along two main therapeutic tracks: (1) unbiased drug repurposing and (2) antisense oligonucleotide (ASO) strategies. Both these strategies are not dependent on patient-derived cell types to commence, but these cells will be critical for follow-up of initial screening efforts. Therefore, we will also address patient cell line acquisition strategies to expedite this growing demand from NFIX researchers.
Other therapeutic modalities need de-risking
Gene therapy approaches such as CRISPR/Cas9-based modalities hold great promise in principle, but will need de-risking and therefore are not considered as a prime focal point in our suggested research activities for the Malan Syndrome Foundation. This modality is currently in very active development in the biotech space, so this decision might be revisited contingent on the outcome of drug repurposing and ASO projects.
Balancing drug-repurposing and precision-medicine approaches
There are many challenges with treating Malan syndrome that necessitate a multi-modality treatment approach. NFIX is expressed in many tissues and Malan patients therefore have clinical presentations affecting different organs in distinct ways. Finally, we cannot predict the deliverability challenges of a novel therapy, especially given that the affected tissues range from the brain to muscle, the retina as well as poorly-vascularized microenvironments such as bone. Hence, the best strategy is to develop multiple modalities in parallel, so that an eventual treatment of Malan might be made up of a combination of different treatment regimes, tailored to the tissue of choice.
Therapeutics readiness
The development of new medicines requires disease models, patient-derived biosamples, and a deep mechanistic understanding of pathophysiology that identifies disease modifying drug targets, including the monogenic driver gene itself. NFIX is a transcription factor whose function is to upregulate the expression of target genes in many cell types, including neuronal and muscle progenitors. Mutations in Malan patients cluster in and around the conserved DNA-binding domain of NFIX (2). Most of these mutations cause an early stop and the mutant mRNA is degraded by nonsense-mediated decay, thereby halving the dose of functional NFIX. Therapeutic efforts must therefore restore the dose of NFIX, either by increasing the expression of the remaining wildtype allele or by promoting its protein functional activity. Accordingly, simple cellular models (patient-derived cells and other tissue-specific cell lines) can demonstrate very well the degree to which a given intervention normalizes, for example, the upregulation of NFIX or its gene targets. Higher animal models then show how haploinsufficiency of NFIX emerges a phenotype, which varies between species and between tissues within a species.
But how much does the particular manifestation in, say, mouse brains, tell us about humans? Divergence of pathophysiology between even closely related species can occur in spite of overwhelming genome sequence conservation. As a result, all information gained from disease models must be multi-factor authenticated and, as such, consensus of mechanism and functional recovery in multiple models is needed to provide the best chance of therapeutic benefit.
Therefore, therapeutic efforts could be focused on 1: Increasing abundance of NFIX in target tissues and 2: Increasing the activity of NFIX at target gene promoters. As a primary readout, we are looking for an increase in protein levels of NFIX or increased activity of NFIX by using an NFIX-specific reporter gene. Protein levels are a more reliable metric than mRNA expression here, as these are directly correlated to the activity of NFIX in the cell. As both an increase in the levels of NFIX protein as well as an increase in NFIX target gene expression are easily assessed in a cell culture setting, simple cellular models of NFIX heterozygosity (either as patient-derived cell-lines or CRISPR/Cas9-engineered generic cell lines) can demonstrate very well the degree to which a given intervention normalizes NFIX protein levels (using antibody staining of native protein or fluorescent protein-coupled NFIX reporter lines) or target gene activity (using luciferase reporter assays based on well-characterized NFIX target gene promoters).
For in vivo follow-up to assess the efficacy across tissues, dose regime and toxicity of an intervention, several in vivo models (mouse, zebrafish, worm) are available to the research community, some of which were developed with support from the Malan Syndrome Foundation. To ensure that potential therapeutics identified in NFIX cellular models translate to in vivo action on NFIX levels or activity, these in vivo models will be the next logical step in identifying an effective therapeutic. Each model system comes with its own set of strengths and weaknesses, which will be outlined in the section below. For the interpretation of drug efficacy results from different in vivo models it is important to keep in mind that the pathophysiology of NFIX haploinsufficiency could differ significantly between even closely related species in spite of overwhelming genome sequence conservation. As a result, our ideal therapeutics candidate would show a consensus mechanism and functional recovery of the NFIX phenotype in multiple models. This type of therapeutics candidate would have the best chance of real-world benefit and would therefore be the most suitable candidate for providing preclinical data for any trials going forward.
Disease Models
Cell-based and animal models of Malan syndrome allow for the experiments necessary to identify potential therapies. We are lucky in that many in vivo models for Malan syndrome have already been developed by the research community and characterized through basic research. However, patient-derived cell-based models to study the diversity of NFIX alleles that give rise to Malan syndrome have not yet been generated and necessitate characterization. We acknowledge this as a major bottleneck for therapeutic development and outline strategies to meet these needs below.
Cell-based models
Patient-derived cell lines are fundamental for investigating therapeutic avenues in treating Malan syndrome. Although commercially-available fibroblast and neuroblast cell lines are being used to model NFIX haploinsufficiency in proof-of-concept experiments, these models are not sufficient for the in-depth characterization needed for therapeutic development. Because Malan syndrome arises from a large spectrum of mutations to the DNA-binding domain of the NFIX gene, we need to understand how drug candidates act to ameliorate the disease phenotype in cell lines derived from many pathogenic NFIX gene variants.
The Malan Syndrome Foundation has efforts underway to build a patient repository of patient-derived peripheral blood mononuclear cells (PBMCs) and fibroblasts for downstream applications such as iPSC generation and cell screening. Although the foundation has a large network of potential sample donors, they have only collected PBMCs from only a few individuals and have no patient fibroblasts available to date. This is because the sample collection is a major challenge for individuals with Malan Syndrome– subjecting donors to a blood draw or biopsy punch have proven to be prohibitively invasive procedures. Alleviating the bottleneck of sample collection, especially for the generation of patient-derived fibroblast cell lines, will be a key step for the development of any and all therapeutic modalities.
To address the bottleneck of increasing donor participation in biobanking efforts, we propose developing a program for the collection of renal-derived stem cells from urine. Urine-derived renal stem cells (USCs) are mesenchymal-like stem cells present in the urine that can be isolated, cultured, and differentiated into UiPSCs (Urinary iPSCs) (4). Remarkably, this is a cost-effective procedure with high yield of UiPSC colonies whose timeline from sample collection to cell line differentiation takes about six weeks. The USCs without any reprogramming can be maintained for more than 50 generations in culture (5,6) and due to their mesenchymal stem cell-like nature, they can be directly reprogrammed to muscle precursors or neuron cell types (5,7) which are clinically relevant models for modeling Malan syndrome.
We have initiated a collaboration with UriCell, a company born out of the laboratory of Prof. James Adjaye at the Institute for Stem Cell Research and Regenerative Medicine at the University of Düsseldorf in Germany. This company specializes in the use of USCs for disease modeling, with an emphasis on kidney disease. They are eager to collaborate with the Malan Syndrome Foundation to generate up to five cell lines from donor samples as a pilot group in an effort to identify costs and streamline protocols for future, larger donor cohorts. A technical hurdle for generating USCs is that it depends on isolation of cells from fresh, unfrozen urine, and therefore necessitates that samples are processed within 24 hours of collection. Because UriCell is located in Germany, we will contact families in the Malan Syndrome Foundation’s network that reside in Europe, which comprise 40% of the contact registry.
We envision the expansion of efforts to collect USCs as a collaboration between the Malan Syndrome Foundation, UriCell, and the CombinedBrain biobank. The Malan Syndrome Foundation already has a working relationship with CombinedBrain for biobanking PBMCs, and because the protocol for generating cell lines from PBMCs, skin samples, or urine-derived epithelial cells follows the same general workflow, we have reached out to Terry Jo Bichell at CombinedBrain and to IBX to discuss efforts for the outsourcing of the urine collection method to IBX as an non-invasive alternative for patient cell biobanking for the Malan Syndrome Foundation and other foundations under the CombinedBrain umbrella. To facilitate this effort, we would need to arrange for the outsourcing of UriCell’s protocol for USC-isolation and biobanking to a US-based partner such as IBX. CombinedBrain is happy to work with either UriCell or an academic partner in the U.S. to set up a pilot for this type of sample collection. They will reach out to IBX in 2-3 months to see if they would be interested in setting up the technology on the U.S. side, in potential collaboration with UriCell.
Though efforts are underway for USC sample collection and cell line generation, we acknowledge that the Malan Syndrome Foundation has not yet solicited skin samples from their network in order to generate fibroblast cell lines. Although a skin biopsy procedure would be prohibitively invasive for many individuals with Malan, we would still encourage the Malan Syndrome Foundation to reach out to their community with a well-formulated appeal for fibroblast donation. This appeal should clearly outline the potential impact of the procedure and weigh this with the potential impact that these samples would have in generating therapeutics screening models and increasing the knowledgebase around Malan syndrome. The availability of multiple patient-derived cell models of different origins will minimize the risk of any cell-type specific artifact that may inappropriately skew screening of a potential therapeutic. Lines derived from multiple cell types from multiple patients (and therefore, many NFIX variants) lays the groundwork for a strong launch into therapeutic development.
Vertebrate models
Vertebrate models of Malan syndrome have been well-developed and characterized for years by leaders in the field such as Prof. Richard Gronostajski and Dr. Matthew Piper who have been SAB Board members for the Foundation. Their work has shown that Nfix/+ heterozygous mutant mice recapitulate many of the key muscular, skeletal and neurological features of Malan Syndrome and therefore could function as a useful study model for preclinical in vivo testing (3,8–10). Currently, efforts are underway to construct a humanized mouse model of NFIX (replacing mouse Nfix with the human gene encoding NFIX) in Dr. Piper’s lab. This model will serve as an even better proxy for the human condition and would be a good entry point both for testing of drug candidates and for optimizing parameters such as efficacy, dosing and delivery method of ASOs targeting NFIX in an in vivo setting. In addition to mouse models, the lab of Dr. Graziella Messina has developed a zebrafish NFIX (nfia) model, which recapitulates some of the muscular defects of the mouse model and could function as another, more economical model for drug testing, especially for initial adverse or toxic effects.
Invertebrate NFIX models
Both roundworms (C. elegans) and fruit flies (Drosophila melanogaster) have a single family member of the NFI family compared to higher vertebrates such as mice and humans, which have four. However, invertebrate model systems do provide the opportunity to study gene function in vivo in a massively parallel way with short turnaround times, especially in the case of C. elegans. The Malan Syndrome Foundation already invested $5000 in the development and phenotypical screening of a C. elegans nfi-1 (NFIX in worms) model and to use their automated motion recording platform and Machine Learning-driven motion analysis to identify motion (i.e. muscle, neuronal) defects in nfi-1 mutant worms. This will be covered in more detail in the Therapeutic Track I section of this document.
Additional model organisms available for NFIX study
NFIX is orthologous to the Nfl gene in Drosophila melanogaster. Single-Cell sequencing of Fly tissues by the FlyCellAtlas consortium (https://flycellatlas.org/) show an enrichment of Nfl expression in muscle tissues of the gut and thorax (flight muscles). Importantly, several genetic tools to both knock down and overexpress Nfl exist in the community that can be ordered from the central Bloomington Drosophila Stock Center in Bloomington, In. These include tools to do tissue-specific Nfl knockdown or CRISPR/Cas9 targeting of Nfl using the well-used Gal4/UAS system. These tools would allow the study of NFIX-like TFs in a tissue-specific manner to further dissect differences in NFIX function in different tissues such as the nervous system, muscle and eye.
Clinical Trial Readiness
The clinical presentation of Malan syndrome offers several qualitative and quantitative metrics for evaluating drug efficacy in a clinical trial setting. The proposed multi-pronged clinical assessment protocol enables the evaluation of drug targeting to several tissue types, such as neuronal and muscle cells.
To evaluate drug efficacy in neuronal and muscle cell types, clinical trialists will continuously monitor changes to sensory impairments and muscle tone that are characteristic of Malan syndrome. Standard audiometry tests (Walker et al., 2013), measures of strabismus (11), and depth perception (12) are routine and non-invasive protocols for detecting improvements to the affected sensory systems. Standardized metrics for measuring hypotonia (13) will be employed to detect any improvements in muscle tone or joint hyperextensibility. The above methods are ideal for use in children with Malan in that they all rely on non-verbal cues for accurate measurement, therefore foregoing the need for verbal communication to obtain reliable clinical data.
Further investigations of drug efficacy in neuronal cell types can be measured by visual field tests and behavioral neurodevelopmental assessments. Automated perimetry quantifies several indices of the visual field to precisely detect any amelioration to baseline defects (14). Finally, the Vineland Adaptive Behavior Scale is recognized by the FDA as a validated instrument for tracking and reporting progress in neurodevelopmental behaviors in response to treatment (15).
Clinical trialists will monitor dosing and efficacy of a repurposed drug or promising ASO at the molecular level by continuously monitoring the upregulation of NFIX target genes in the urine-derived cells of individuals with Malan syndrome. The same renal stem cells obtained from urine and cultured for drug development (4) will be collected over the course of a clinical trial to directly and non-invasively assess the regulation of NFIX targets by RT-qPCR.
The clinical endpoints enumerated in this section will be modified throughout the Roadmap project. Specifically, the data generated during Year One activities will serve as the basis of a pre-Investigational New Drug (IND) meeting request submission. We will receive responses from FDA on what exact safety measures and clinical assessments will be needed for a Malan Syndrome experimental therapy.
Standardized Clinical Trial Protocol
The experiments performed during Year One of this Roadmap will build a foundation for a pre-Investigational New Drug (IND) meeting request for the treatment of Malan Syndrome. Feedback from the pre-IND meeting will give us clarity on what kind of clinical study will best serve patients in the development of therapies.
Therapeutic Tracks for Malan Syndrome
The knowledgebase around NFIX and Malan syndrome has expanded significantly over the last 10 years and the effects of the molecular lesions in NFIX on the Malan phenotype are well-defined in most cases. Based on the literature, Malan Syndrome is due to a haploinsufficiency of the NFIX gene, where the DNA binding activity of protein being made from one copy of the NFIX gene is abolished. Therefore, any therapy that would increase either the levels or activity of the wild type NFIX would hold great promise for successfully treating Malan Syndrome. Since NFIX is also part of a family of transcription factors that have overlapping binding specificities, it is possible that a drug that modulates activity of any of the NFI family members could mitigate haploinsufficiency of NFIX. Furthermore, there may be yet-unidentified factors in tissues whose compensatory mechanisms may be revealed by an unbiased drug repurposing screen.
Our strategy for finding the first Malan medicine has two main recommendations:
Track I: Drug repurposing
Track II: precision medicine ASO approach
While both approaches would benefit from the availability of patient-derived cell models, initial proof-of-concept experiments are underway in generic cell lines that express NFIX. Hence, efforts to increase the availability of patient cell lines for the community and therapeutics efforts should run in parallel. Below we will outline each therapeutic track and give recommendations for future directions.
Therapeutic Track I: Drug repurposing
Drug repurposing is defined as identifying a new use for an already approved drug, be it a century old drug or a drug that was just recently approved. This is an attractive option for multiple reasons. First of all, FDA-approved drugs won’t need to be de-risked for use in humans to the same degree as novel compounds. This will significantly cut development timelines for new treatments using an already FDA-approved drug to target a rare disease such as Malan. Lastly, many of these drugs are produced commercially and are available at a fraction of the costs of a novel compound (16). We think that the short timelines and cost-efficient nature of drug repurposing screens should make this one of the key strategies for finding a possible therapy for Malan syndrome. Below we will outline the drug repurposing strategies that should preferably be run in parallel to find a suitable drug for boosting NFIX levels or activity.
In vitro drug repurposing screen using a luciferase-based NFIX reporter gene
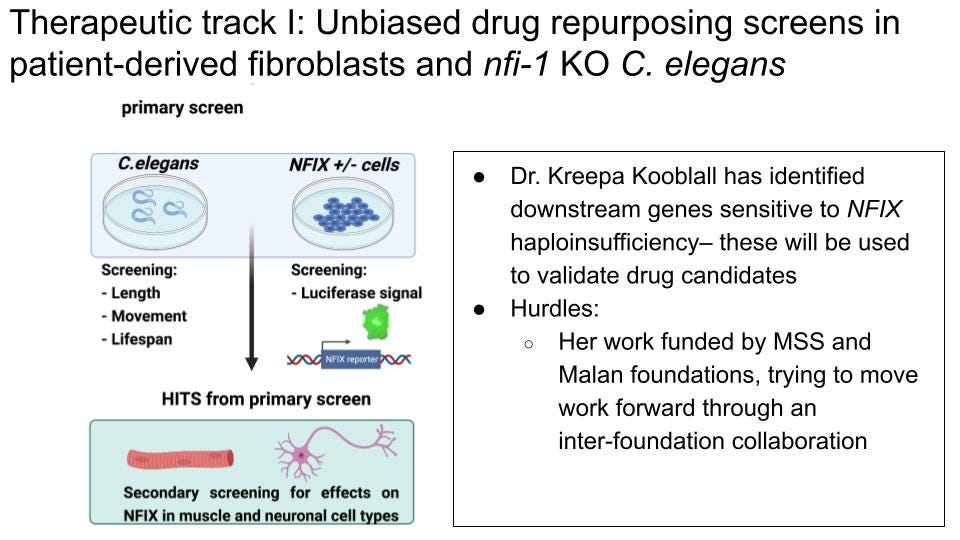
A reporter assay for an unbiased drug repurposing screen has already been developed by Dr. Kreepa Kooblall in Rajesh Thakker’s Group at Oxford University. They generated a construct that contains an endogenous NFIX-responsive promoter to drive luciferase expression in fibroblasts as a readout for observing changes in NFIX activity. Although the NFIX-responsive promoter element’s activity needs to be validated in target tissues (namely, the brain and muscle), a luciferase assay is attractive because it yields a quantitative readout of NFIX binding to DNA. The amplitude of the luciferase signal would be higher in cells with two copies of the wildtype NFIX gene compared to those with only one wildtype copy. This assay is therefore amenable to a high-throughput drug repurposing screen, where a compound that increases the amplitude of the luciferase signal is considered a candidate for restoring the “dose” of NFIX binding. We would recommend first optimizing this reporter for maximum amplitude when comparing its activity in WT, WT/NFIXDel and NFIXDel/NFIXDel genotypes. We would need to create stable reporter cell lines for each of these genetic conditions to be able to use these cells for drug-repurposing screening. These would need to be commercially generated through a CRO such as Synthego, Ubigene or Expresscells and could be generated for around $10,000-15,000 total. We have reached out to several of these companies to gather quotes for the work.
Dr. Kooblall’s work has also identified downstream target genes of NFIX. These will serve as secondary markers for decreased NFIX activity in the follow-up on drug candidates identified in the primary luciferase-based screen and can be used to validate activity of the drug in different tissues in an in vivo model further down the line. Changes in mRNA and protein abundance of these downstream targets will thus be used to validate “hits” in the primary screen. The work of Dr. Kooblall will therefore lead to an efficient NFIX activity reporter as well as a validated set of secondary markers for any follow-up screening with drug candidates in relevant in vitro tissue models such as muscle and neurons.
This work to date has been funded by a grant from the MSS Foundation. As both Malan and MSS are caused by loss of NFIX activity, we would urge a pooling of resources for the financing of a drug repurposing screen based on Dr. Kooblall’s luciferase assay and NFIX target genes. A single drug repurposing screen for both syndromes would be the most efficient and economical way for these foundations to push this approach forward and achieve actionable therapeutic candidates for further in vivo studies using patient-derived cells and the Nfix mouse models available in the community. Our recommendation would be to enlist Charles River Laboratories for this screen, since they have a very well-developed high-throughput screening facility that uses the extensive Broad Repurposing Hub drug library. To drive this forward, we have already reached out to the MSS Foundation and have set up a meeting to discuss options for future collaborations in this therapeutic track. The Malan Syndrome Foundation has also established contact with the CACNA1A-Foundation, and recurrent microdeletions around chromosome 19p13.2 affects both the CACNA1A and NFIX gene. Hence, some CACNA1A patients might also be affected by NFIX mutations. Therefore, investments from all three foundations would benefit therapeutics efforts for all three patient groups.
In vivo drug repurposing screen using C. elegans
A drawback of the luciferase reporter assay is that it uses a single promoter element to model a representative NFIX binding site, which is unlikely to model all variations in the genomic environments that might influence NFIX activity. To address this challenge, we propose an independent drug repurposing track that measures NFIX activity in vivo in a target-agnostic manner. Though the assays we propose in this section are separate from Dr. Kooblall’s work and its ties to the MSS foundation, we invite inter-foundation collaborations to pool resources and supercharge drug screening efforts.
Next to work in cell culture models, the Malan Syndrome Foundation has also been working closely with InVivo Biosystems to develop a C. elegans model that is suitable for a drug repurposing screen. To date, the company has generated very compelling evidence that nfi-1 mutants have a screenable phenotype that correlates with the clinical presentation of Malan syndrome. High-content phenotyping revealed nfi-1 mutant worms have a significantly altered mean amplitude of body bending when moving and an altered body length. Both movement phenotypes and altered body length match phenotypes seen in Malan patients. Furthermore, Day 4 adult nfi-1 worms assumed a very distinct “infinity” pose “∞” that was virtually not seen in the control and therefore constitutes a unique screenable phenotype for any drug repurposing screen. We recommend moving this effort forward with either InVivo Biosystems or Magnitude Biosciences. This U.K.-based company also has experience with high-throughput image screening of large collections of chemical and aging interventions. Another option for drug-repurposing screens in C. elegans is the lab of Dr André Brown at Imperial College London. Perlara is partnering with Dr. Brown’s lab for another rare disease drug-repurposing project and Dr. Brown has years of experience in automated screening of movement phenotypes for both research and commercial partners. Alternatively, Vivaltes Therapeutics in the Netherlands is an C.elegans-specialized drug screening firm run by Dr. Marjolein Wildwater, who is a former colleague of Dr. Korzelius, one of the authors of this Roadmap. Before moving forward with this worm model though, we would urge validation of the nfi-1 deletion used: (i) Confirmation that the deletion results in a null mutation with no detectable mRNA for nfi-1 or if the phenotype still produced a truncated version of the NFI-1 protein that carries the transactivating domain; (ii) Testing of relevant phenotypes not only in the nfi-1 homozygous deletion, but also in a nfi-1/+ heterozygous worm, which would be a better match for the Malan syndrome genetics.
The movement phenotype is a clinically-relevant phenotype for Malan syndrome, but also one which could yield a treatment for a variety of muscle-degenerative disorders. Whereas haploinsufficiency for NFIX leads to hypotonia and affects gait of Malan patients, work from Dr. Graziella Messina’s lab has shown that NFIX loss can actually lead to improved muscle function in a model of Duchenne muscular dystrophy (DMD) (17). Hence, this presents an opportunity to kill two birds with one stone: drug candidates that upregulate NFIX levels or function are candidates for Malan and MSS Syndrome treatment, whereas molecules that inhibit NFIX would be candidates for the treatment of a range of muscular dystrophy disorders. While the focus for the Malan Syndrome Foundation should lie with strategies that screen for upregulated NFIX levels/activity, the link between NFIX and muscular dystrophy treatment presents an opportunity to tap into a much larger disease community. Therefore, our recommendation would be to establish contacts with these communities, possibly through Dr. Messina, to partner up in further developing suitable readouts for NFIX levels and activity. The muscular dystrophy disorder space is a much larger disease space for drug development, with significant investments from both new and established pharmaceutical companies. Building the case for NFIX as a targetable molecule for both the treatment of Malan/MSS and muscular dystrophy disorders would make large-scale investments into treatments targeting NFIX a much more attractive strategy for these companies.
Cellular models for follow up of primary screen candidates
Selected drug candidates from efforts described above will need to be tested in relevant target tissues. To this end, we suggest two main cell types for follow-up: the muscle and Neural Progenitor Cells. Both cell types play key roles in the Malan Syndrome pathology and strategies are available to derive these cell types from a range of patient samples: blood, urine or skin fibroblasts, as outlined in the “Cell-based models” section of this document.
Muscle: For the follow-up of primary screen candidates, there are several CROs offering high content screening of muscle cell types. Cytoo (Boston, MA and Grenoble, France) specializes in the high-content screening of skeletal muscle and cardiomyocytes phenotypes. Alternatively, Ncardia (Leiden, The Netherlands) is a company that specializes in iPS-derived cardiomyocyte models for drug screening efforts. The starter cell types for this would be primary human myoblasts or iPSCs, respectively, both derived from Malan patients. These cells would be acquired through UriCell and can be differentiated into the relevant cell types by these companies. As Dr. Messina’s work has clearly shown significant effects of Nfix mutation on mouse myoblast and skeletal muscle function (18,19), this type of cellular model would be a good candidate for follow-up of primary screen hits.
Neurons/Neural Progenitor Cells (NPCs): As Malan Syndrome patients show several neurodevelopmental phenotypes, a secondary cell-based model of relevance to Malan would preferably be neuronal. Work from Dr. Gronostajski and Dr. Piper have delineated the various neurological phenotypes associated with Nfix loss of function using mouse models of Nfix heterozygosity that mimic many Malan syndrome phenotypes. Nfix loss leads to overproliferation of neural Stem Cells (NSCs) in Culture (20) as well as differentiation defects of adult Neural Progenitor Cells (NPCs) in vivo (3,8,10). Cell cycle quiescence in NSC is easily detected using standard assays such as Ki67 staining or EdU/BrdU incorporation. Therefore, the effect of any drug candidate from the primary screen on NSC-quiescence using these standard assays would be a low-risk follow-up experiment. Both methods to establish NSCs from iPSCs as well as BrdU/EdU and Ki67 staining are commercially available and well-established and these experiments could be performed easily by both CRO-partners such as Charles River or academic labs.
Therapeutic Track II: Precision medicine/ASO
Precision medicine can be defined as a therapy that uses a patient’s specific genetic or molecular profile to prevent, diagnose, or treat a disease (National Cancer Institute). In the case of Malan syndrome, this would mean developing strategies to upregulate the levels of the NFIX gene directly. This is a good strategy for risk mitigation, as hits from the drug repurposing track might influence the expression of the NFIX luciferase reporter gene directly through NFIX or through a parallel genetic pathway that acts on the NFIX target gene. In case of the latter, targeting of a parallel pathway might lead to off-target effects. Therefore, a precision medicine strategy that focuses on directly regulating NFIX levels will be essential to develop further in the long-term strategy of the Foundation. The main therapeutic approach we will explore in this section is a variant-specific targeting strategy using antisense oligonucleotides (ASOs) whose efficacy can be evaluated within the coming 6-12 months. Another strategy we propose will focus on gene editing as a back-up long-term plan for treating Malan syndrome in case our first precision medicine strategy fails.
Upregulation of the wild-type NFIX allele
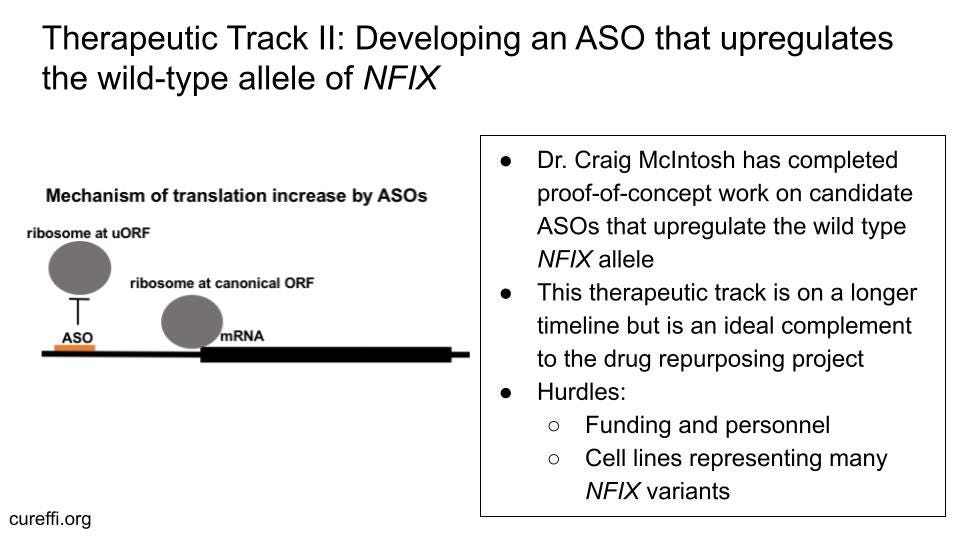
Because individuals with Malan syndrome have one functional allele of the NFIX gene next to the defective one, therapies that upregulate the expression of the correct allele to correct the haploinsufficiency are exceptionally attractive treatment avenues. Antisense oligonucleotide (ASO) therapies regulate the availability of an mRNA transcript, resulting in changes to the abundance of a protein (and protein variants) of interest. These strategies are relatively variant-agnostic, as most variants of NFIX in Malan Syndrome patients are in the coding exon 2 of NFIX and the ASO strategy outlined below targets the 5’ Untranslated Region (UTR), which is unaffected in most Malan Syndrome patients.
The Malan Syndrome Foundation granted $30,000 to developing an ASO that upregulates the healthy NFIX allele to Dr. Craig McIntosh, a postdoctoral researcher in May Aung-Htut and Steve Wilton’s research group at the Centre for Molecular Medicine and Innovative Therapeutics at Murdoch University. Their strategy is to target the upstream Open Reading Frames (uORFs) in the 5’ UTR of the wild-type NFIX allele to increase the levels of functional NFIX protein. Upstream ORFs or uORFs are small regulatory elements upstream of the primary ORF (in this case: NFIX) that negatively regulate expression from the primary ORF (21,22). Targeting these elements with ASOs can be an effective therapeutic strategy for upregulation of genes in several genetic diseases (23,24). Dr. McIntosh presented compelling proof-of-concept data at the Malan Syndrome Scientific Roundtable in December of 2021 demonstrating that this is a promising therapeutic approach for correcting the haploinsufficiency that causes Malan.
The research on NFIX led by Dr. McIntosh shows exceptional promise in light of the group’s previous work in developing FDA-approved ASO-based therapeutics for other rare diseases. Dr, Aung-Htut and Dr. Wilton both work with Sarepta Therapeutics, where Dr. Wilton has licensed several FDA-approved treatments for Duchenne Muscular Dystrophy and both researchers are pursuing the same drug licensing and approval strategy for the treatment of Pompe disease. Given the group’s proven track record for drug development in conjunction with Dr. McIntosh’s carefully-executed research activities, we propose continuing to further fund the group’s efforts towards developing an NFIX-specific ASO.
With additional funding, Dr. McIntosh's efforts will narrow down which candidate ASO sequences, concentrations, and chemistries are the most effective in upregulating the abundance of the functional NFIX protein. Although the proof-of-concept work was conducted in commercially-available SHSY-5Y neuroblastoma cell lines, this next stage of ASO development necessitates more clinically-relevant models of Malan syndrome. Dr. McIntosh plans to collaborate with Dr. Piper at the University of Queensland for in vivo validation of final ASO candidates in a mouse model of Malan syndrome. In order to identify final ASO candidates to test in vivo, Dr. McIntosh has emphasized the need for patient-derived cell lines that represent a wide array of pathogenic NFIX variants. Our plan is to make the iPSCs that are currently being generated available to the groups as soon as they are available, which will hopefully allow for the next step of ASO development while we are developing the urine-derived stem cell collection program for generating a library of UiPSC lines.
In parallel to the strategy of upregulating wild type NFIX through ASO-mediated uORF manipulation, we propose exploring partnership opportunities with Stoke Therapeutics, a biotech company that specializes in ASO therapies specifically designed to treat haploinsufficiency. Their Targeted Augmentation of Nuclear Gene Output (TANGO) technology is specifically tailored to increase expression of a given allele by down-regulating the targeting of transcript for nonsense-mediated decay (NMD), rather than increasing transcriptional output from the gene promoter. We would first need to reach out to Stoke Therapeutics to ask if this strategy will be applicable to NFIX, since not all genes are amenable to this ASO-strategy. However, pursuit of two different gene-specific and variant-agnostic ASO-based therapeutic strategies would ensure a comprehensive exploration of therapeutic avenues which increases the likelihood of timely development of treatments for the Malan community writ large.
Gene therapy using CRISPR/Cas9-based base editing techniques
Another strategy for correcting the affected allele in Malan Syndrome patients is the use of CRISPR/Cas9 technology to correct the affected exon 2 of NFIX. Although CRISPR/Cas9 holds great promise for precision medicine, various technical factors make this more of a long-term strategy in our opinion. First, whereas CRISPR/Cas9 technologies have been successful to cure e.g. sickle cell anemia (Vertex : CTX001), these therapeutic strategies rely on manipulating cells ex vivo in a cell culture environment before replacing them in the host. This type of strategy is not suitable for a neurodevelopmental disorder such as Malan, since neuronal and muscle cells cannot be explanted and replaced at will. In vivo CRISPR-strategies are mainly based on the use of adenovirus-derived AAV vectors delivering the gene editing payload in a target tissue of choice. Although AAV vector-based therapies are well tolerated and several are in clinical trial stage, delivery strategies to neuronal tissues have been shown to have highly variable efficiency (25). Recent advances such as CRISPR/Cas9 based base-editing hold great promise towards correcting nucleotides in vivo without Cas9 nuclease activity that could result in second site mutations and triggering of the DNA damage response in target tissues (26). However, the main challenge will still be how to deliver the respective Cas9-based therapeutic to its target tissue, especially if multiple tissues are involved such as in Malan syndrome. It is worth noting however that this is a very rapidly changing space and the playfield of many start-up biotech firms that hold great potential for driving this type of therapeutic track to maturity. Hence, this space deserves watching to identify future partnerships with start-ups that could deliver a gene therapy track for Malan syndrome to pre-clinical stage readiness.
Research Portfolio management
Next to the activities described above, the Malan Syndrome Foundation has several other projects and partnerships ongoing to advance their goals of treatment and finding a cure for Malan syndrome. We will outline these efforts in the section below and make recommendations on strategies for moving forward.
Partnerships and alliances with other foundations
The Malan Syndrome Foundation has done an impressive job in establishing links with other Foundations that have overlapping interests in studying NFIX specifically and links to other overgrowth syndromes more generally. In the case of NFIX, specifically, there are historic links with the Marshall Smith Syndrome (MSS) Research Foundation. As outlined above in the Therapeutic Readiness section, the fact that both syndromes are allelic disorders caused by alterations in NFIX should be clear grounds for deepening collaboration efforts between both Foundations. Perlara has been helping to revitalize these ties in order to expedite drug repurposing screens targeting NFIX. We have a meeting set up to discuss options for mutual financing and fundraising to support these efforts and hope to help the Malan Syndrome Foundation guide these efforts in the future. In addition, the Malan Syndrome Foundation has been working on founding an “Overgrowth Alliance”, focused on syndromes with different genetic causes, but with many overlapping symptoms, such as overgrowth, megacephaly, ataxia and different types of seizures.
The Foundation has contacts with Prof. Kate Tatton-Brown, a specialist in overgrowth conditions and co-discoverer of the overgrowth condition Tatton-Brown-Rahman Syndrome (TBRS) (27,28). Further candidates for inclusion in this Alliance would be Smith-Kingsmore Syndrome Foundation (autosomal dominant MTOR mutations), Beck-Fahrner syndrome Foundation (autosomal dominant and recessive TET3 mutations) and the Sotos Syndrome Foundation (autosomal dominant NSD1 mutations). To kickstart this idea, we would suggest following the successful example of the Malan Roundtable Meeting. This type of event would bring together all relevant stakeholders and identify mutual targets for research, treatment options and fundraising efforts. This will diversify the knowledgebase of the Malan Syndrome Foundation and expand their network of researchers and clinicians interested in these types of syndromes. Perlara would be able to help by identifying and contacting key stakeholders as well as in the organization and moderation of the Round Table event. By focusing on similar phenotypes instead of a similar affected gene, this presents an alternative approach to find treatment options for Malan-associated symptoms. In addition, this Alliance will open up a broader base for fundraising and generating interest from the pharmaceutical industry for addressing the shared symptoms in these conditions.
Additional research efforts on NFIX and its regulation
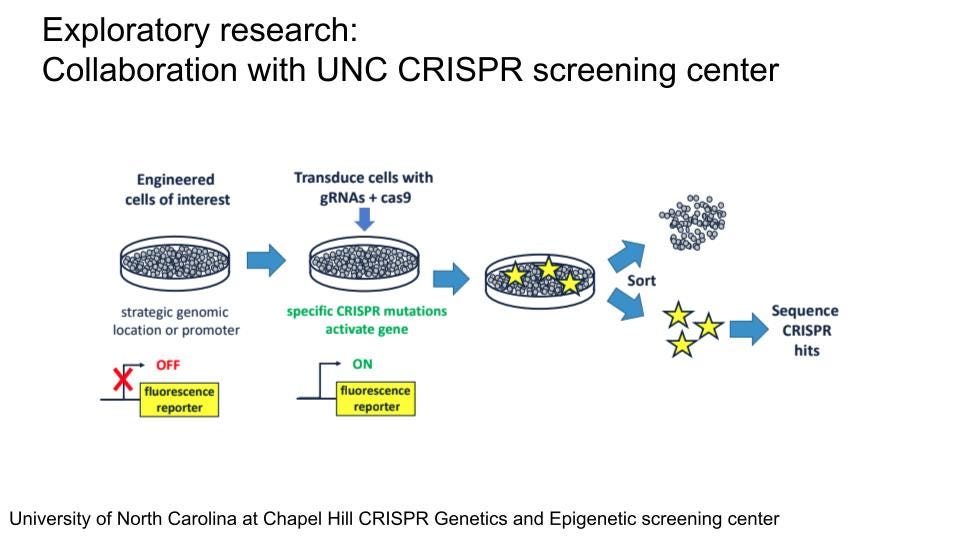
Apart from the efforts described in the Therapeutic Readiness section, several other efforts are underway to better understand NFIX and its regulation. The Malan Syndrome Foundation has funded research by Prof. Erin Heinzen at UNC Chapel Hill to establish an iPSC-derived model of Malan syndrome and studying the effects of the NFIX mutation in neuronal differentiation and function. In addition, there is a plan to perform CRISPR-based pooled screening at the UNC CRISPR Screening Facility for factors affecting NFIX levels on cell lines expressing a fluorescent protein-tagged NFIX (NFIX-FP) reporter. Both efforts have been held back by technical issues and the fact that the UNC CRISPR Facility has just been established. Generation of both genetically modified iPSCs and fluorescently tagged NFIX-FP cell lines for CRISPR-screening has been slow due to limited capacity at the UNC Stem Cell Core on which they are reliant. Timelines were suggested of up to one year, which would unnecessarily further delay this screening effort. Unfortunately, there seems to be a reluctance of commercial providers such as Synthego to provide the custom services needed for this project. The use of patient-derived iPSCs from urine would again be a useful alternative for generating NFIX heterozygous iPSCs and a more faithful model for studying NFIX function in neuronal function and development. The CRISPR Facility seems to be still under active development and reliant on UNC sources for the generation of the NFIX-FP reporter. As the generation of these cell lines is the main obstacle at this time point, we would suggest a more extensive search of possible companies beyond Synthego and identification of academic partners that would be willing to take over the generation of this cell line in an effort to move things forward and to check in regularly on the progress made at UNC with these projects. However, once established, this approach holds great potential for the identification of new, potentially druggable pathways which affect NFIX abundance.
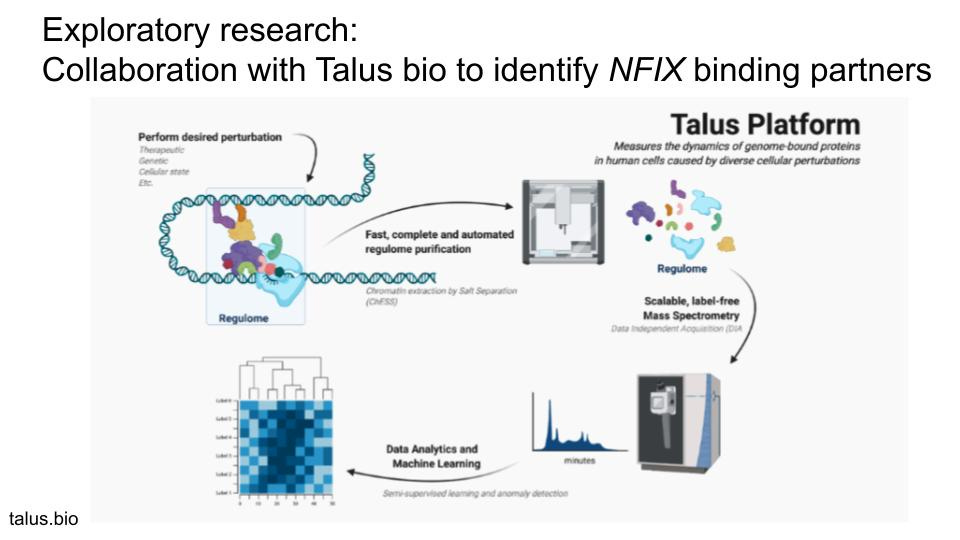
Understanding NFIX and its “Regulome” the TalusBio platform for studying transcriptional regulators
NFIX performs its role as a transcription factor (TF) in the cell by regulating transcription of specific genes in different cell types of the body. TFs seldom do this alone, but often have a number of associated proteins that are part of a so-called “regulome” that helps regulate the transcription of its target genes or bind the TF to inhibit its function, depending on the cellular state and context. At Perlara, we have connections with a biotech start-up called Talus Bio that uses a proprietary platform named Marmot tailored for identifying the regulatory network of TFs as they bind to their target DNA in the cell and how this binding changes when a cell is treated with a certain drug. Not only will this platform be able to identify the “regulome” associated with the NFIX TF, it can also be used to test the ability of different drugs in stabilizing or destabilizing the NFIX regulome in the cell. We have established contacts with Alex Federation, Co-founder and CEO of Talus Bio, who is willing to take on NFIX as a showcase study for their platform’s ability to transform rare disease research associated with DNA-binding factors such as NFIX. We believe that this provides an opportunity to contribute to both drug discovery and knowledgebase efforts around NFIX for the Malan Syndrome Foundation.
References
1. López-Rivera JA, Pérez-Palma E, Symonds J, Lindy AS, McKnight DA, Leu C, et al. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain. 2020 Apr 1;143(4):1099–105.
2. Priolo M, Schanze D, Tatton-Brown K, Mulder PA, Tenorio J, Kooblall K, et al. Further delineation of Malan syndrome. Human Mutation. 2018 Sep;39(9):1226–37.
3. Piper M, Gronostajski R, Messina G. Nuclear Factor One X in Development and Disease. Trends in Cell Biology. 2019 Jan;29(1):20–30.
4. Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012 Dec;7(12):2080–9.
5. Bharadwaj S, Liu G, Shi Y, Markert C, Andersson K-E, Atala A, et al. Characterization of Urine-Derived Stem Cells Obtained from Upper Urinary Tract for Use in Cell-Based Urological Tissue Engineering. Tissue Engineering Part A. 2011 Aug;17(15–16):2123–32.
6. Pavathuparambil Abdul Manaph N, Al-Hawwas M, Bobrovskaya L, Coates PT, Zhou X-F. Urine-derived cells for human cell therapy. Stem Cell Res Ther. 2018 Dec;9(1):189.
7. Rahman MS, Wruck W, Spitzhorn L-S, Nguyen L, Bohndorf M, Martins S, et al. The FGF, TGFβ and WNT axis Modulate Self-renewal of Human SIX2+ Urine Derived Renal Progenitor Cells. Sci Rep. 2020 Dec;10(1):739.
8. Oishi S, Harkins D, Kurniawan ND, Kasherman M, Harris L, Zalucki O, et al. Heterozygosity for Nuclear Factor One X in mice models features of Malan syndrome. EBioMedicine. 2019 Jan;39:388–400.
9. Campbell CE, Piper M, Plachez C, Yeh Y-T, Baizer JS, Osinski JM, et al. The transcription factor Nfixis essential for normal brain development. BMC Dev Biol. 2008 Dec;8(1):52.
10. Harris L, Dixon C, Cato K, Heng YHE, Kurniawan ND, Ullmann JFP, et al. Heterozygosity for Nuclear Factor One X Affects Hippocampal-Dependent Behaviour in Mice. Borlongan CV, editor. PLoS ONE. 2013 Jun 11;8(6):e65478.
11. Mohney BG, Holmes JM. An Office-Based Scale for Assessing Control in Intermittent Exotropia. Strabismus. 2006 Jan;14(3):147–50.
12. Birch E, Petrig B. FPL and VEP measures of fusion, stereopsis and stereoacuity in normal infants. Vision Res. 1996 May;36(9):1321–7.
13. Soucy EA, Wessel LE, Gao F, Albers AC, Gutmann DH, Dunn CM. A Pilot Study for Evaluation of Hypotonia in Children With Neurofibromatosis Type 1. J Child Neurol. 2015 Mar;30(3):382–5.
14. Flammer J, Drance SM, Augustiny L, Funkhouser A. Quantification of glaucomatous visual field defects with automated perimetry. Invest Ophthalmol Vis Sci. 1985 Feb;26(2):176–81.
15. de Bildt A, Kraijer D, Sytema S, Minderaa R. The psychometric properties of the Vineland Adaptive Behavior Scales in children and adolescents with mental retardation. J Autism Dev Disord. 2005 Feb;35(1):53–62.
16. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019 Jan;18(1):41–58.
17. Rossi G, Bonfanti C, Antonini S, Bastoni M, Monteverde S, Innocenzi A, et al. Silencing Nfix rescues muscular dystrophy by delaying muscle regeneration. Nat Commun. 2017 Dec;8(1):1055.
18. Messina G, Biressi S, Monteverde S, Magli A, Cassano M, Perani L, et al. Nfix Regulates Fetal-Specific Transcription in Developing Skeletal Muscle. Cell. 2010 Feb;140(4):554–66.
19. Rossi G, Antonini S, Bonfanti C, Monteverde S, Vezzali C, Tajbakhsh S, et al. Nfix Regulates Temporal Progression of Muscle Regeneration through Modulation of Myostatin Expression. Cell Reports. 2016 Mar;14(9):2238–49.
20. Martynoga B, Mateo JL, Zhou B, Andersen J, Achimastou A, Urbán N, et al. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 2013 Aug 15;27(16):1769–86.
21. Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proceedings of the National Academy of Sciences. 2009 May 5;106(18):7507–12.
22. Barbosa C, Peixeiro I, Romão L. Gene Expression Regulation by Upstream Open Reading Frames and Human Disease. Fisher EMC, editor. PLoS Genet. 2013 Aug 8;9(8):e1003529.
23. Crooke ST, Liang X-H, Baker BF, Crooke RM. Antisense technology: A review. Journal of Biological Chemistry. 2021 Jan;296:100416.
24. Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012 Nov;69(21):3613–34.
25. Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. 2018 Sep;17(9):641–59.
26. Huang TP, Newby GA, Liu DR. Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat Protoc. 2021 Feb;16(2):1089–128.
27. Yokoi T, Enomoto Y, Naruto T, Kurosawa K, Higurashi N. Tatton-Brown-Rahman syndrome with a novel DNMT3A mutation presented severe intellectual disability and autism spectrum disorder. Hum Genome Var. 2020 Dec;7(1):15.
28. Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, et al. The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 2018 Apr 23;3:46.