

Pharma Chameleon
Often hiding in plain sight, a small molecule can have multiple pharmacological identities. Context-switching and shape-shifting pharmacophores keep showing up as hits in our drug repurposing screens.


Here’s a pharmacological riddle wrapped in a biological conundrum: what do two plant-based natural products that rescue the growth of yeast and worm models of a pediatric inborn error of metabolism have in common with a human-made drug approved in 1992 in Japan for adults with diabetic neuropathy?
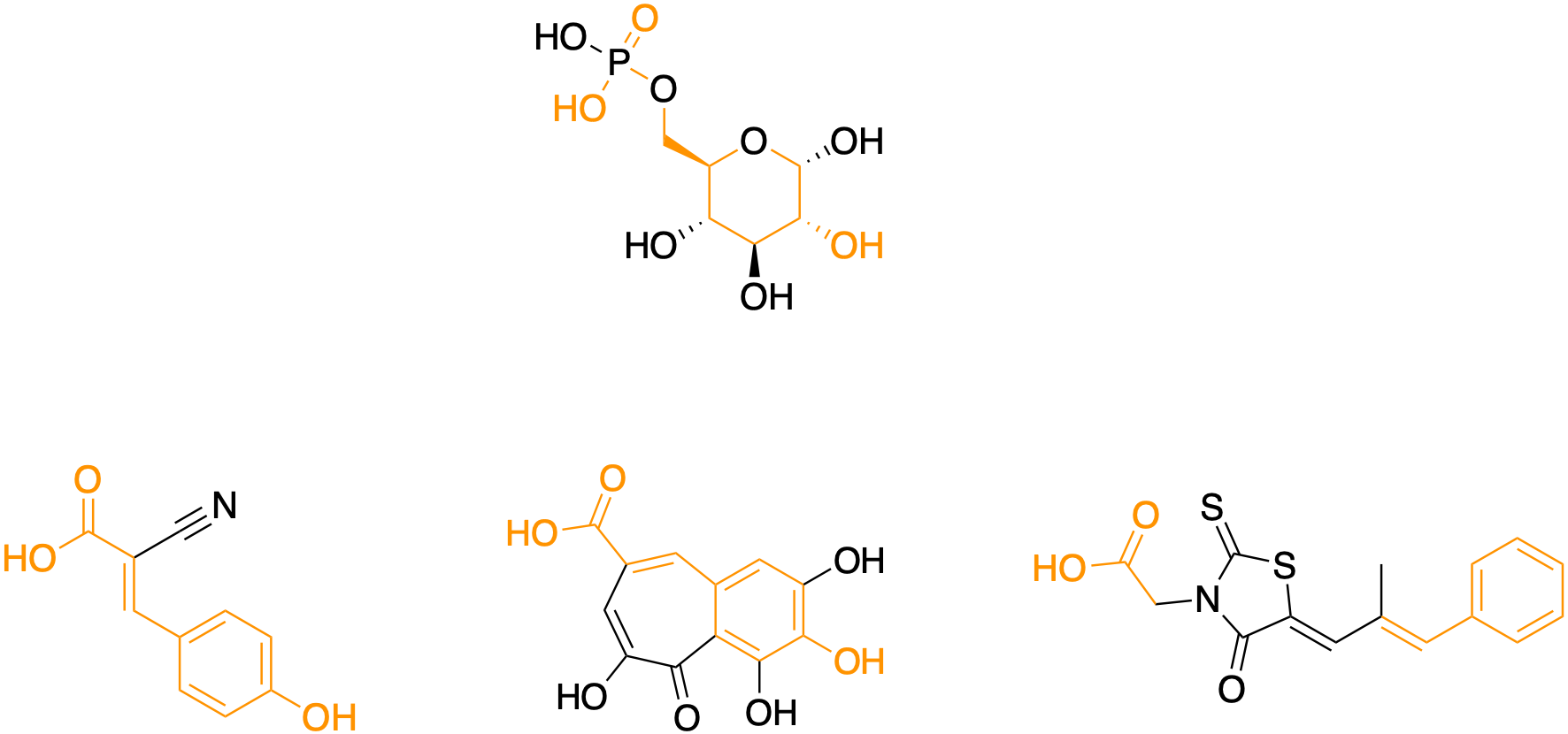
The carboxylic acid containing pharmacophore highlighted in orange is a strong hint, indicating the existence of a flexible and spacious binding site in a conserved drug target present in yeast, worms and humans. That still leaves hundreds of suspects.

Need to use a lifeline? One of the phytochemicals, alpha-cyano-4-hydroxycinnamic acid (CHCA), is the active component of the beloved spice cinnamon, which is reported to have anti-diabetic (insulin mimicking) properties in addition to its undisputed flavor enhancement. The other phytochemical, which is isolated from the Plume Poppy tree, is also a hydroxycinnamate and its name is purpurogallin-4-carboxylic acid.
Notice that CHCA can be superimposed on purpurogallin-4-carboxylic acid. In CHCA, the carboxylic acid has some degree of conformational flexibility at the end of the propylene side chain. On the other hand, the carboxylic acid of purpurogallin-4-carboxylic acid is locked in place by the cycloalkene ring. The diabetes drug has an ungainly rhodanine heterocycle wedged in between the carboxylic acid and phenyl ring.
Together, the structure-activity relationships paint a picture of a buried binding site — more like a divet — for the carboxylic acid near the surface of the target protein. Meaning the carboxylic acid is the business end of the molecule. It must form specific hydrogen bonds with positively charged amino acids that line the active site of the target protein. Requirements for the shape and size of the backside of the molecule are loose.
Still stumped? Okay, here’s a final clue. The synthetic drug is called epalrestat. It is still branded as Kinedak in Japan. It was originally developed by Ono Pharmaceuticals and approved in Japan in 1992 as a treatment for peripheral neuropathy in adults with Type 2 diabetes (T2D).
If you haven’t miraculously guessed it yet, the answer to the pharmacological riddle is that all three compounds are aldose reductase inhibitors (ARIs). Aldose reductase is an evolutionarily conserved enzyme that converts glucose to sorbitol. Aldose reductase is the first committed step in the polyol pathway. Chronic polyol accumulation inside cells is toxic. Sorbitol in particular instigates the peripheral neuropathy that is a painful and life-limiting part of T2D.
Now that we’ve ostensibly solved the pharmacological riddle, let’s turn our attention to the biological conundrum. Why does an ultra-rare congenital disorder of glycosylation (CDG) caused by a hard-wired deficiency of an indispensable sugar-modifying enzyme have anything to do with insulin resistance and its sequelae aka T2D, a common polygenic disease of aging influenced by diet and lifestyle choices?
Put another way: how does aldose reductase inhibition unite CDG and T2D? The resolution to the biological conundrum is that too much glucose is a bad thing. That creates a paradox at the heart of metabolism. Food is both fuel and poison; both life-giving and intoxicating, depending on the dose and exposure. It all comes back to metabolic balance.
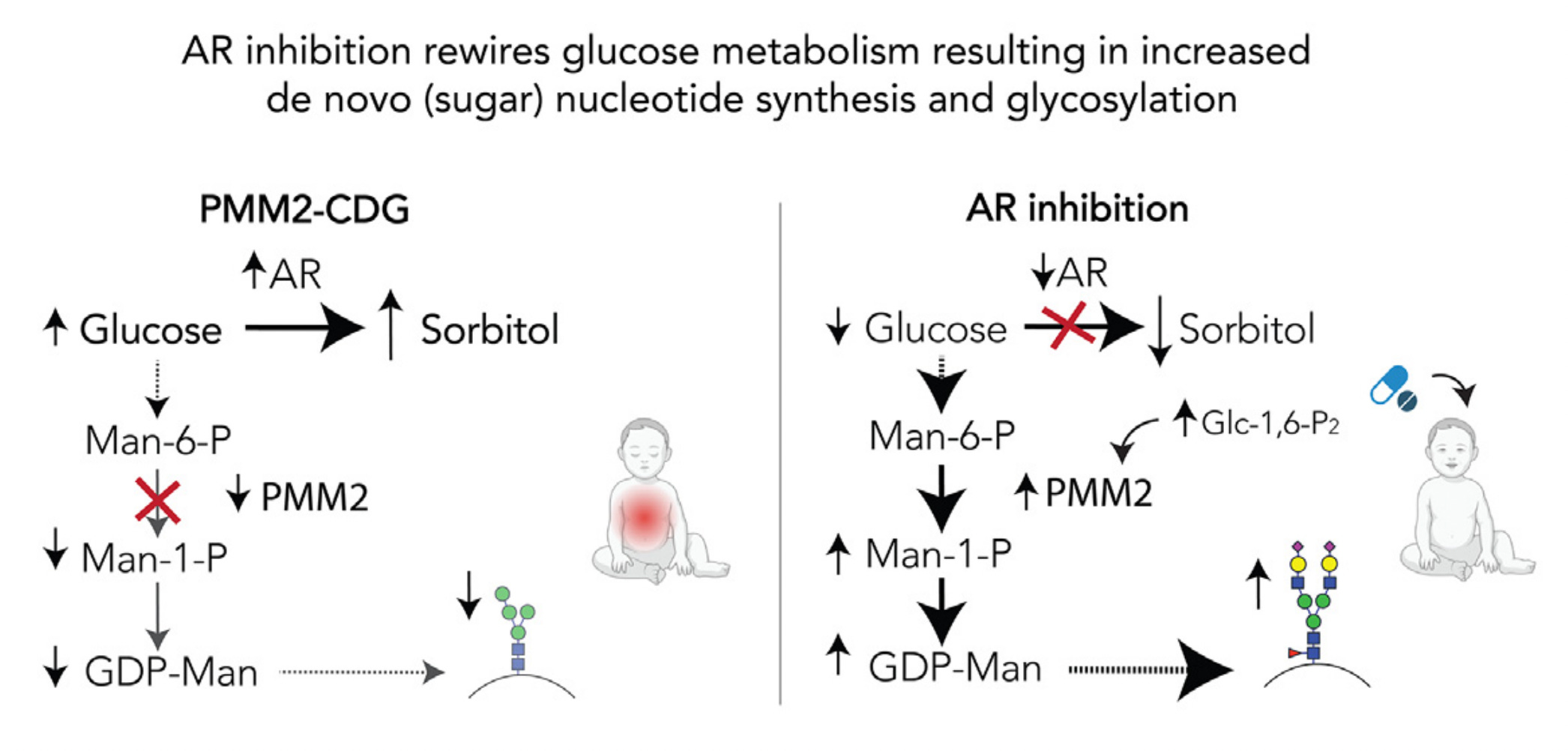
Life evolved a compromise that involves temporary polyol production as a pressure valve to deal with sudden unexpected surges in glucose levels. But prolonged exposure to excess glucose — as happens with an inborn error of metabolism and a chronic metabolic disease of adulthood — polyol production becomes a lesser-of-two-evils means by which cells cope with the stresses and consequences of excess glucose.
The inability to fuel N-linked glycosylation means that every cell in the body experiences a tiny excess of internal glucose that is no longer being used to make functional proteins, but has to go somewhere. In the case of adult-onset diabetes, cells and tissues of the body are bathed in a tiny excess of dietary glucose. Whether from without or from within, the levels of glucose are too high. Too much glucose is a multi-layered complex disease driver.
We made those leaps of logic by staring at chemical structures of hits from yeast or worm powered phenotypic screens. In other words, courtesy of OI: Organic Intelligence. (No AI model can do this today). We merrily plunged headfirst down a pharmacological rabbit hole until we burrowed into the aldose reductase active site. From there, we backed into epalrestat because it was the only commercially available and clinically actionable aldose reductase inhibitor.
Peering into the vastness of chemical space, we can’t help but wonder what other disease biology connections and multi-purpose drug targets are hiding in plain sight. When using patient avatars as a lens, we see chemical space in an entirely new light. Like being able to perceive parts of the spectrum that were previously obscured or invisible. Or like discerning once-camouflaged creatures in the forest for the first time.

Epalrestat is currently being evaluated for safety and efficacy in children living with PMM2-CDG as part of a pivotal Phase III trial at Mayo Clinic. What follows is a summary of the many steps that got us here. Full disclosure: Maggie’s Pearl is the commercial sponsor of the Phase III trial, and Perlara is a co-owner of Maggie’s Pearl.
Longtime followers will know that the PMM2-CDG PerlQuest was launched in the beginning of 2017. The first yeast models of PMM2-CDG were created that summer. In the beginning of 2018, we completed drug repurposing screens with yeast PMM2-CDG avatars. That yeast campaign yielded three rescuer hits, including the cinnamon compound CHCA. The same drug repurposing library was screened on a worm model of PMM2-CDG in the summer of 2018. That worm campaign yielded 20 hits, none of which are the same as the three yeast hits. However, two of the three yeast hits are plant-based polyphenols, and so are the majority of the worm hits.
Once we appreciated the family resemblance between the two carboxylic acid containing yeast and worm hits, it wasn’t long before sleuthing on PubMed led us to aldose reductase as a key protein of interest. After beholding the chemical structure of epalrestat for the first time, the pieces of the puzzle came together, in 2D at least. Aldose reductase was a “hot target” back in the 1990s given the size of T2D market (even back then), so there are many crystal structures available online. Including crystal structures with bound ligands and inhibitors!
A flattened representation of the aldose reductase active site and the contours of the ARI pharmacophore are shown below. As predicted, an anion binding pocket is lined by a pair of neighboring tryptophan and histidine residues. The other two defining features of the ARI pharmacophore are a specificity pocket and a hydrophobic pocket.

To really make the case, we have to go 3D. Below is a rendering of an epalrestat fragment containing a V-shaped carboxylic acid warhead that projects out of the plane of the rhodanine scaffold. The nicotinamide moiety of the the NADP+ cofactor is floating above and off to the side of epalrestat.
Here’s the identical perspective but now the amino acid side chains have been filled in along with hydrogen bonds indicated by the dashed lines. (Water molecules are the red dots). The carboxylic acid of epalrestat is hydrogen bonded to the active site triad of histidine, tryptophan and tyrosine.
What about the rest of the molecule besides the carboxylic acid? A space-filling model of the aldose reductase protein is shown below on the left. NADP+ is the green molecule that inserts its nictotinamide group into the protein. In magenta are 10 different aldose reductase inhibitors that were co-crystalized with aldose reductase. The carboxylic acid warhead also inserts into the submerged active site. Remarkably, all of the aldose reductase inhibitors orient their carboxylic acid groups in the same position in space. So long as the carboxylic acid group is moored to the active site by the catalytic triad, there are no structural impositions on the rest of the molecule.

There’s one last twist. The story gets more satisfying when you realize it’s a double cipher. There may be second, deeper answer to the riddle that explains the connection between two different diseases of metabolism that affect humans at different stages of life. It also may explain why so many plants evolved a pharmacopiae of anti-diabetic natural products in the first place.
Shape-shifting molecules with multiple embedded (and in some cases overlapping) pharmacophores all target enzymes in the same metabolic pathway because of substrate mimicry. Take a closer look above at the compound rendered in black in superimposed aldose reductase inhibitors. It’s none other than glucose-6-phosphate, which sits atop central metabolism. Sugar-modifying enzymes have an affinity for sugars. It’s obvious in retrospect but it wasn’t obvious at the beginning. The amazing part is that we observe this “takes one to know one” phenomenon in almost every drug repurposing project.
Here’s a second completely independent example of a convergence between substrate mimicry, evolutionary pharmacology and drug repurposing. We’ll switch metabolic gears for this section and discuss a mitochondrial disease.
Complex I deficiency has been treated with ubiquinone dietary supplementation, most commonly as CoQ10. The ubiquinone supply chain is fueled by essential precursors called polyprenols. What if there was a way to stimulate ubiquinone production inside cells versus delivering it from the outside via diet? Are there factors that promote endogenous ubiquinone synthesis in a Complex I deficient cell whether a yeast or a human neuron?
Complex I deficiency can arise from mutations in dozens (if not hundreds) of genes. Our standby Saccharomyces cerevisiae is not a great fit because it lacks a functional Complex I. So we chose a yeast species with a functional Complex I and an obligate aerobe (requiring oxygen for life) like humans: Yarrowia lipolytica.

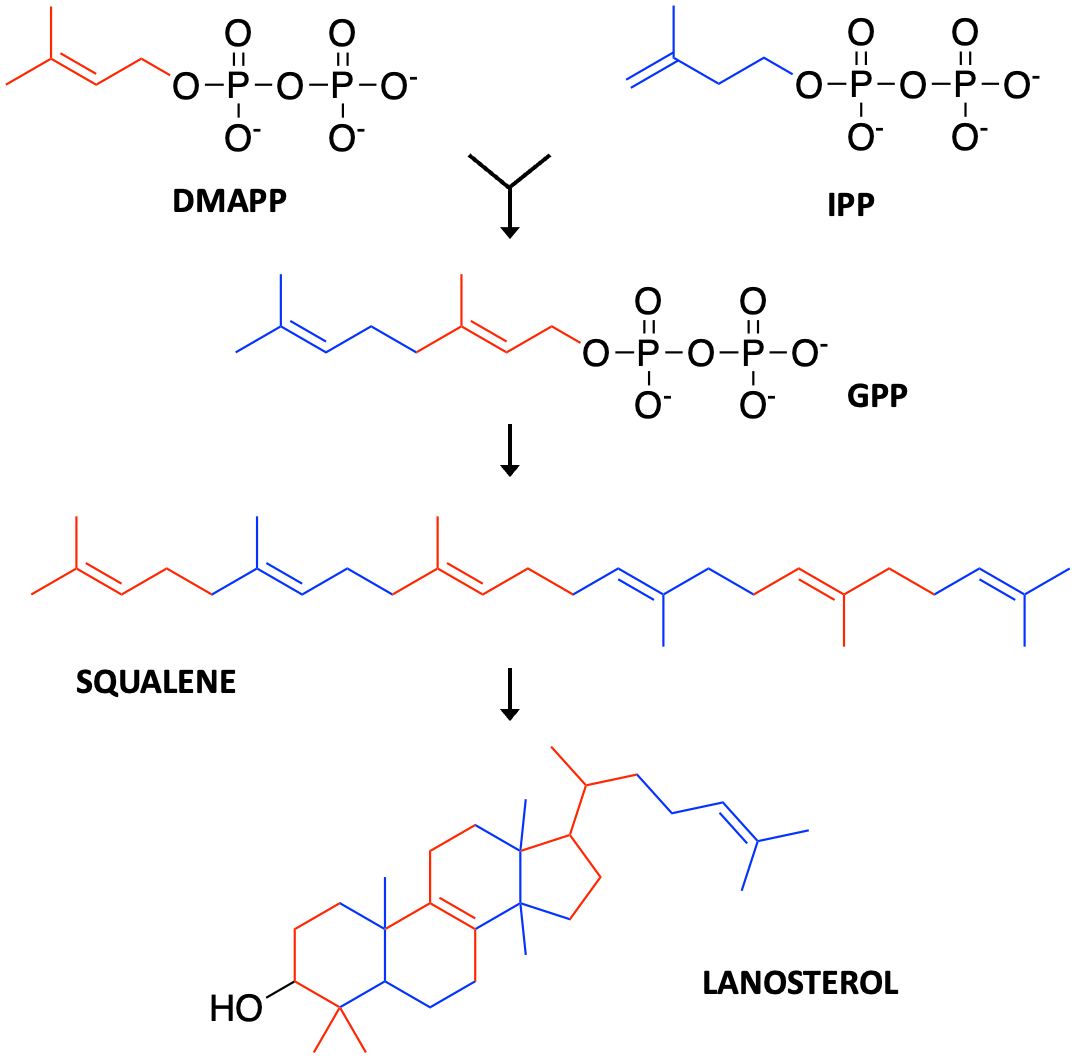
The simplest and cheapest was to create a Complex I deficiency yeast avatar is to expose wildtype cells to a Complex I natural product toxin called piericidin A, which happens to look a heckuva lot like ubiquinone. Substrate mimicry strikes again! This time it’s isoprene subunits, which you can think of as Lego bricks. CoQ10 has ten isoprene subunits snapped together in a row, forming a flexible tail that runs down the barrel of Complex I.
Yarrowia lipolytica yeast cells were treated with piericidin A and grown either in glucose media or in acetate media for up to 84 hours or 108 hours, respectively. Four polyprenols — parthenolide, nerolidol, bisabolol and farnesol, natural product polyprenol precursors — are strong rescuers of piercidin A toxicity. Whereas geranylgeranoil, linalool, citronellal, menthol and limonene are inactive at the concentration screened.
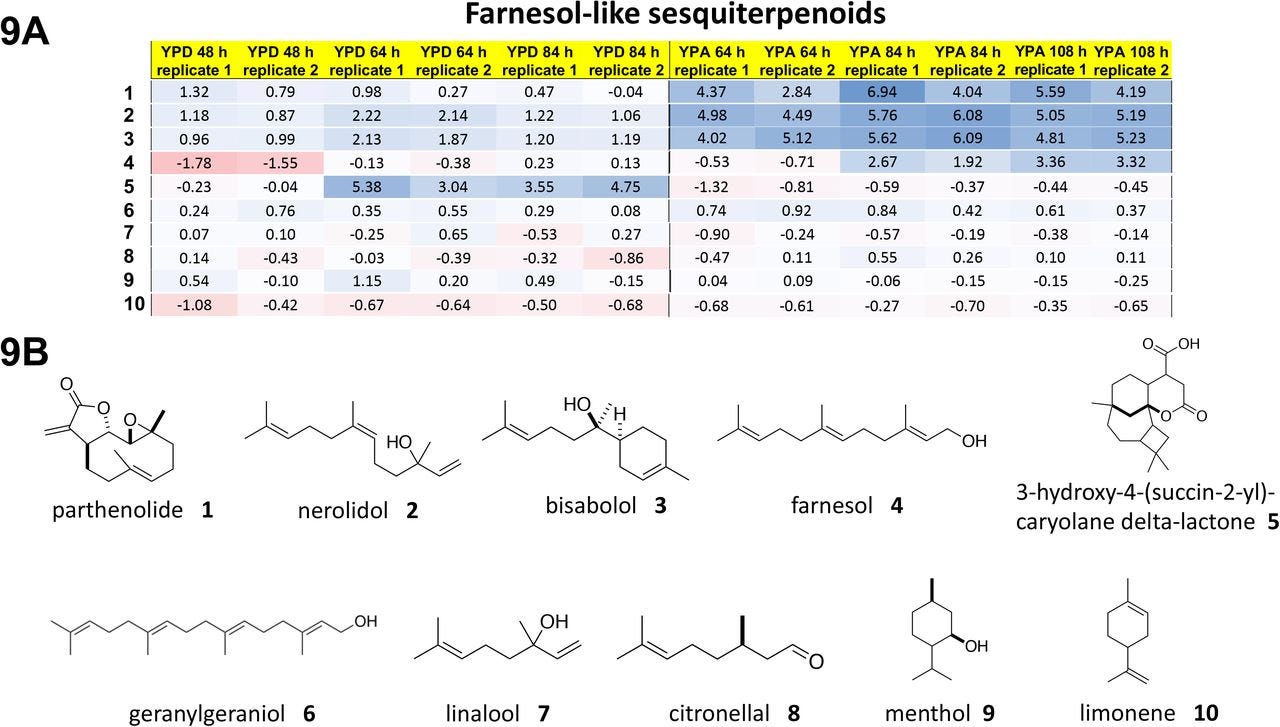
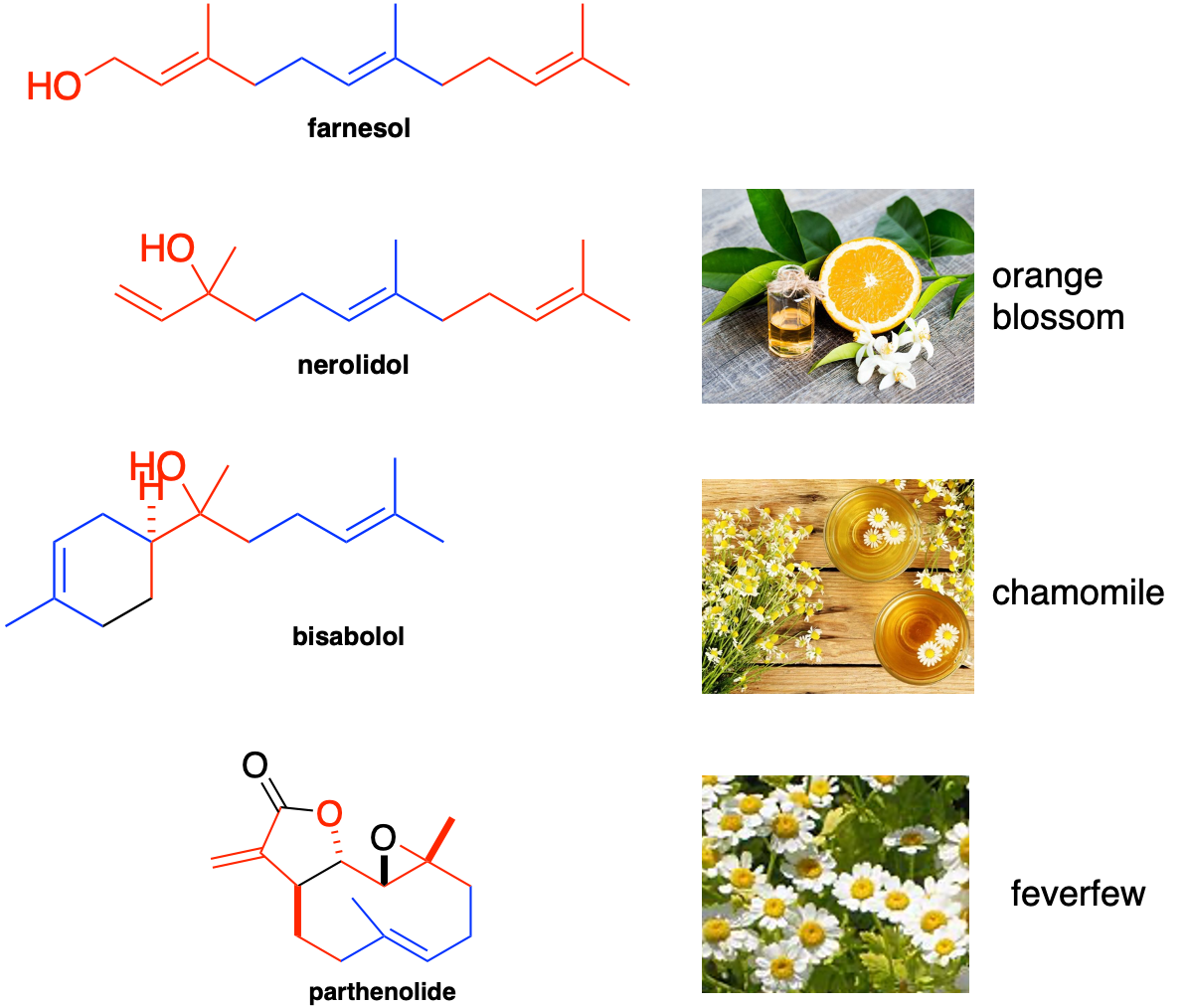
What pops out right away is that piericidin A, ubiquinone and the rescuer hits are all polyprenols (in contrast to the polyphenols in the preceding section). The sesquiterpene farnesol pharmacophore is evident below in all four rescuers. The shape-shifting ability of the farnesol pharmacophore is on glorious bichromatic display. It also just so happens that farnesol, which the cell supercharges to farnesol pyrophosphate, is at the chokepoint in mevalonate metabolism that feeds down toward ubiquinone production.
The inactive compounds tell a consistent story. Geranylgeraniol is a diterpene and so has one extra isoprene Lego brick. It is downstream of farnesol and located after the ubiquinone spur, so it would not be expected to stimulate endogenous ubiquinone production. Linalool and citronellal are aliphatic monoterpenoids lacking an isoprene compared to nerolidol, which is a diterpenoid, and not able to be assimilated into the ubiquinone biosynthetic pathway. Citranellal also has the reactive aldehyde moiety. Menthol and limonene are cyclized monoterpenes.
Don’t believe your eyes? The other major branch downstream of mevalonate flows to cholesterol biosynthesis. Just like farnesol can be twisted and coiled into parthenolide, squalene is folded up into lanosterol in much the way a clown makes a poodle out of a balloon at a birthday party.
Once we knew what to look for, substrate mimicry crops up all over our drug repurposing hit lists. The first example is glucose-6-phosphate. The second example is farnesol. A third example is mevalonate, a stone’s throw metabolically speaking from either farnesol or glucose-6-phosphate. We previously wrote about this disease (SRD5A3-CDG) and the pioneer family advancing toward a 1-to-N study.
The six SRD5A3-CDG worm hits are united by a mevalonate pharmacophore highlighted in purple.

The presence of atorvastatin immediately put the target of statins, HMG-CoA reductase, on our radar. Atorvastatin clearly extends a mevalonate side chain from its core scafford. The other compounds don’t contain a mevalonate. Or do they?
Mevalonate exists in two forms: acyclic and cyclized. Looking more closely at the anticoagulant dicoumarin, the cyclized form of mevalonate is clearly embedded in the structure, as a doublet no less. Substrate mimicry strikes a third time! Yeast obviously aren’t able to clot since they not only don’t have blood, they don’t have platelets. That means dicoumarin is moonlighting with a second target that is present in yeast.
Plant-based polyphenols do lots of things in cells and animals. We observed plant-based polyphenols in the first example of PMM2-CDG. What’s interesting is the contrast between the two CDGs. SRD5A3-CDG polyphenols are structurally distinct from PMM2-CDG polyphenols. For starters, the SRD5A3-CDG polyphenols contain many hydrophobic methoxy groups in the place of hydrophilic hydroxyl groups that are decorate the PMM2-CDG polyphenols. The SRD5A3-CDG polyphenols in fact contain a partial mevalonate because they are missing the carboxylic acid. Even so, there is literature supporting that these polyphenols can inhibit HMG-CoA reductase. Interestingly, just like the twinning observed in dicoumarin, hypocrellin A is also a doublet.
To conclude, in almost every patient-avatar-powered drug repurposing screen we’ve performed for an inborn error of metabolism we observe a unique substrate-mimicking pharmacophore. Not just any old substrates. We’re talking the most hyper-connected metabolites in cells across evolution, such as glucose-6-phosphate and mevalonate. In an example we will share soon, we even found a small molecule that is active in two different diseases by two different mechanisms. After all these years, it feels like we’re just getting started.
Next steps: 1-to-N studies..