The PLOD1 thickens
After pivoting to biochemical approaches and moving operations from popup lab to a PLOD1 expert's lab at UCSF, we're close to making the go/no go decisions we presented in the PLOD1-kEDS Cure Roadmap.


In collaboration with

When patients are the platform, the last stop on a diagnostic odyssey is the embarkation point for a cure odyssey. In other words, it’s the patients in pursuit of modalities instead of modalities chasing after patients. Perlara’s distributed biotech consultancy model empowers pioneer families to pursue multiple therapeutic paths at once culminating in single-patient trials for their kids that immediately pave the way for the early adopter families in their communities.
We call this framework 1-to-N medicine.
Because patients are the platform, our distributed biotech consultancy model must be designed to be both modality agnostic and disease agnostic. Meaning we have to be prepared to work on any disease or gene or variant. Case in point: PLOD1-kEDS is an ultra-rare type of Ehlers-Danlos Syndrome (EDS). Collectively, mutations in collagen biosynthesis, assembly and maturation genes like PLOD1 are not rare. All types of EDS may be as prevalent as 1 in 5,000 individuals worldwide.
Collagen is a heterotrimeric molecule composed of two α1 chains and one α2 chain, forming an uninterrupted triple-helical structure. The procollagen molecule is cleaved at specific sites at both N- and C-terminal regions. Lysine modifications of collagen are catalyzed by LH1 and the other two lysyl hydroxylase isozymes, leading to the final step of collagen biosynthesis: covalent intermolecular cross-linking to stabilize and rigidify the triple helix.

Inside the cell, specific lysine residues are hydroxylated to form hydroxylysine. Hydroxylysine residues located in the helical domain of the molecule are then glycosylated. Outside the cell, lysine and hydroxylysine residues in the N- and C-telopeptides can be oxidatively deaminated to produce reactive aldehydes that undergo a series of non-enzymatic condensation reactions to form covalent intra- and intermolecular cross-links.
In our last update two months ago, we had just made the handoff of the patient’s fibroblasts to Prof Yoshihiro Ishikawa at UCSF, an expert in LH1. LH1 stands for Lysyl Hydroxylase 1. It is the enzyme encoded by the PLOD1 gene. The patient is homozygous for a 1-basepair deletion in exon 5 that leads to a frameshift and a premature stop codon.

A truncated protein lacking both glycosyltransferase and hydroxylase activities is predicted to be made if mutant mRNA transcripts are not destroyed by nonsense-mediated decay, a quality control system cells have in place that prevents mutant proteins with premature stop codons from being produced.

Up until this point, we still didn’t know what was happening with LH1 and collagen in the patient’s skin cells. Finally, we got some answers last week.
Longtime Cure Odyssey readers will recall that the PLOD1-kEDS CureMap co-authored by Dr Victoria Blake and Dr Shiri Zakin in September 2021 recommended a three-part precision medicine strategy:
An ASO-based exon-skipping therapy to mask the mutation-harboring portion of the gene and restore protein functionality.
A gene therapy delivered by non-integrating virus in somatic (not germline) cells to correct the mutation in the PLOD1 gene.
An enzyme-replacement therapy to deliver functional LH1 to key organs.
Of course we also advocated for drug repurposing to identify compounds that could bypass LH1 or activate one or both of the other two lysyl hydroxylases in the cell (compensatory isozyme activation) or rescue collagen biosynthesis by a novel mechanism we haven’t imagined.
In order to rule in or rule out specific approaches, we had to establish assays to measure LH1 function and collagen properties in the patient’s fibroblasts. Sequencing data confirmed the presence of 1-basepair deletion frameshift mutation in homozygosity in the patient’s fibroblasts. Sequencing data from the paternal fibroblasts is still pending. The pathogenic PLOD1 variant results in a premature stop codon downstream at LH1 amino acid position 227. The full-length wildtype LH1 protein has 729 amino acids.
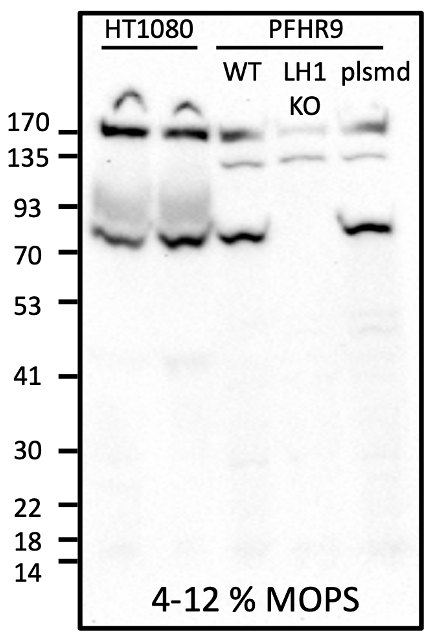
Using a differentiated epithelial mouse cell line (PFHR-9) where PLOD1 was knocked out by CRISPR/Cas9 as a negative control and a wildtype human fibroblast line (HT1080) as a positive control, Yoshi performed Western blot analysis to measure the total amount of LH1 protein in each sample.
As shown below, a band of the expected size was detected between 93-70 kDa in HT1080 and PFHR9. We know the LH1 antibody is truly specific for LH1 because the LH1 band disappears in the LH1 CRISPR knockout cell line. Going one step further, expressing PLOD1 on a plasmid normalizes LH1 expression levels. In concept, we proved that a gene therapy approach could work in mouse skin cells.
A baby step to be sure, but a milestone nonetheless.
Knowing that the negative and positive controls behaved as expected in both human and mouse skin cells, Yoshi then assessed LH1 protein expression in control fibroblasts (C) versus the patient’s fibroblasts (P). Quite strikingly, no LH1 band was detected in the patient cell lysate. In this experiment, an LH1 antibody targeting an epitope (part) of the LH1 protein that is located after the patient’s premature nonsense mutation.

The absence of detectable LH1 make senses, but it doesn’t rule out that a truncated (shortened) version of LH1 is produced in the patient’s fibroblasts. To make sure we weren’t being fooled by the first antibody, Yoshi tested a second LH1 antibody that targets an epitope located before that patient’s premature nonsense mutation. That way if a truncated LH1 protein were produced in the patient’s cells, it would be detectable by this second antibody.
Focus on the band between 93-72 kDa in the duplicate C lanes. Notice that the band disappears in the duplicate P lanes. So we got the same result using both LH1 antibodies: no LH1 band detected in the patient cell lysate.

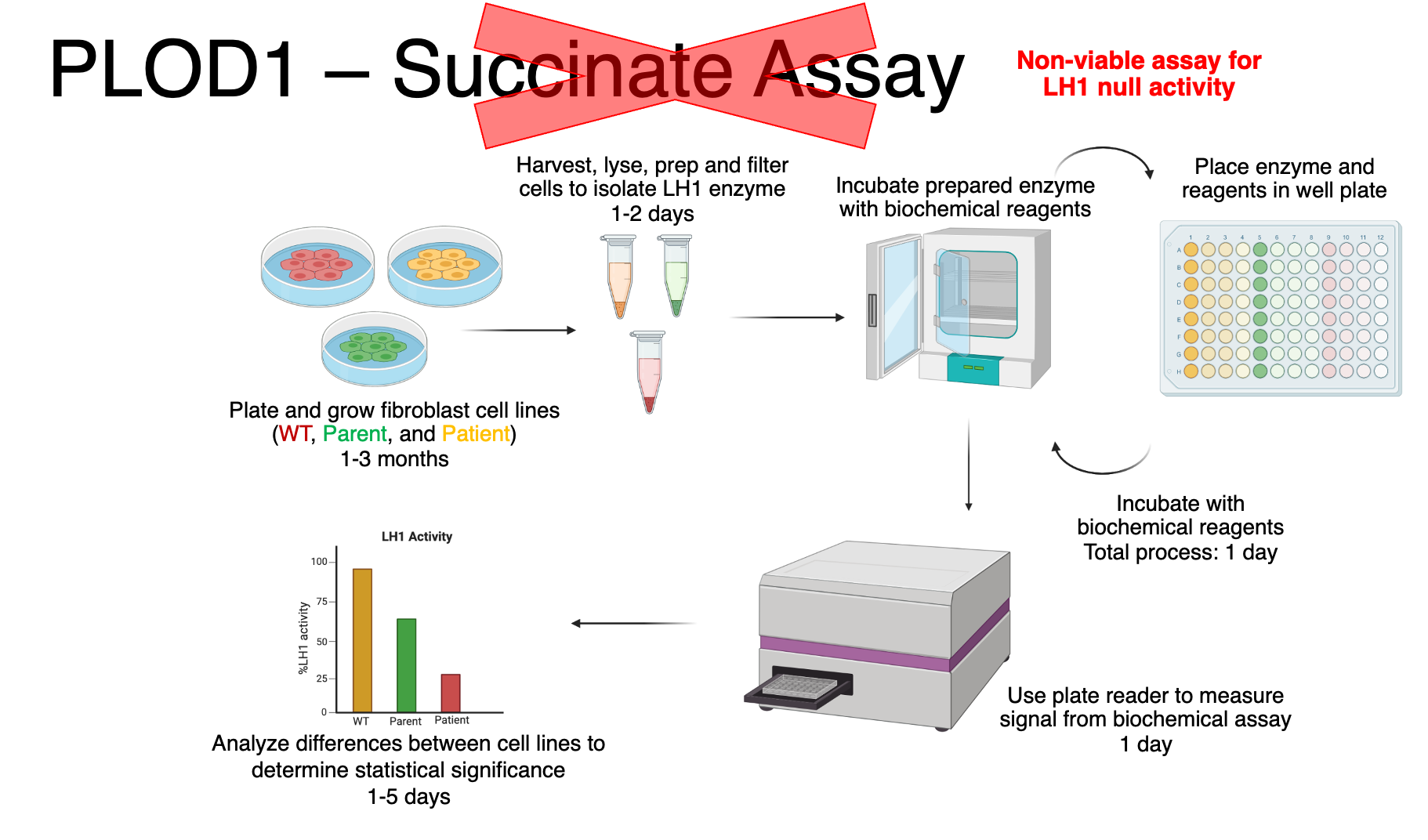
Based on these data, we can conclude that the patient’s PLOD1 frameshift mutation is a total loss-of-function at the protein level and it behaves like a PLOD1 knockout. Also, we can rule out the succinate assay for drug repurposing since there is no detectable LH1 protein in the patient’s cells.
After assessing PLOD1/LH1, we next interrogated collagen biosynthesis inside the patient’s fibroblasts and the structural integrity of collagen secreted into the media by the patient’s fibroblasts. Type 1 collagen (Col1) is detected outside of the cell in cell culture media. As shown below, two bands of roughly equal intensity are detected in the control (C) duplicates, while only one intense higher molecular weight band is detected in the patient (P) duplicates.
Clarifying experiments are planned but our running hypothesis is that terminal cleavage of pro-collagen is not performed in the patient’s cells due to improper lysine hydroxylation, resulting in an abundance of premature collagen (the intense higher molecule weight band) versus mature collagen.

Interestingly, Type 3 collagen (Col3) is detected in patient and control at similar levels. Maybe there’s a modest reduction in the patient (P) samples.

However, Type 5 collagen (Col5) behaves appears significantly reduced in the patient’s cells. If we corroborate this Western blot result in an immunohistochemistry experiment by staining cells for Type 5 collagen and demonstrating a reduction in Type 5 collagen abundance, we could have the rescuable phenotype we need for a high-throughput drug repurposing screen.

Taken together, we hypothesize that various types of collagen rely on lysine hydroxylation via LH1 to varying degrees, resulting in some types of collagen being more affected than others.
Here are the next steps in the project Gantt chart:
Repeat experiments on the paternal cell line and controls: confirm heterozygosity via DNA sequencing
Compare collagen (Types 1, 3, and 5) in patient cells versus the LH1 knockout cell line
Design protease assay to investigate the formation of proper 3-dimensional collagen structures
Run qPCR to see if LH1 mRNA is present to examine if ASO is a viable strategy

Finally, the totality of the results indicate that the ELISA assay for detection of hydroxylysine is still viable. Mouse LH1 knockout lines still show a considerable amount of hydroxylysine present due to some compensation by other LH enzymes. The first set of optimization experiments are being planned as we speak.