

We have PIGA-CDG hits! Now what?
We finally completed the Pharmakon screen of ~2,250 repurposable drugs. We identified 9 hit compounds including an approved drug and an over-the-counter supplement that point to a common target: iron.


In collaboration with



Disclaimer
The results of the PIGA drug repurposing project that we are sharing in the spirit of open science below are novel preclinical research findings and therefore they do not constitute the practice of medicine. Please consult your physician or clinical care team if you’re considering off-label use of any approved drug. The same caution applies to nutraceuticals, supplements and “generally recognized as safe” compounds.
In the previous update from a month ago, we anticipated being able to proceed to the Pharmakon screen without additional assay optimization holdups or hiccups. It’s taken us much longer than we expected to get here because the PIGA gene has been uniquely challenging to model in the lab, even in a single-cell organism like yeast.
Why? The PIGA gene rests on a knife's edge, genetically speaking. Loss of 50% of PIGA function is tolerated without disease, otherwise PIGA-CDG parents/heterozygous carriers would be sick. But complete loss of PIGA is embryonic lethal in all animals including humans.
Disease burden is suffered specifically by boys because of a cruel twist of fate: the PIGA gene happens to reside on the X chromosome. As we all learned in primary school biology class, boys have just one copy of every gene on the X chromosome. Therefore, only variants (whether de novo or inherited from a mom or dad) that preserve residual PIGA enzymatic activity support life in boys.
PIGA mutations observed in boys fall on a spectrum of clinical severity from infant mortality to survival to adulthood. There’s a diverse and unusual constellation of affected organs. Sometimes there’s neurological involvement or facial dysmorphia, sometimes not. Collectively, those observations are consistent with the actions of multiple disease modifiers, which can be either genetic or environmental. Modifiers can make disease progression milder or more severe.
The early essentiality of the PIGA gene, its location on the X chromosome, and the complex interactions between PIGA variants and disease modifiers conspire to create a sphinx-like disease. Despite those unique challenges, we pulled off the first-ever drug repurposing screen for PIGA-CDG — and we got hits. Remarkably, the top hit reinforces a clinical finding from 2013 in one of the first papers to describe boys with PIGA mutations and a new disease called Ferro-Cerebro-Cutaneous syndrome — later renamed PIGA-CDG.
What’s even more exciting, the top PIGA hits have similar mechanisms of action as the top hits from other drug repurposing screens of disease models of congenital disorders of glycosylation (CDGs), including PGAP3-CDG.
As we reported last month, the temperature-sensitive PIGA mutant behaved as expected in the initial seeding density experiment. Next, we performed a Z’ optimization experiment in order to demonstrate that the negative controls (mutant cells) and the positive controls (wildtype cells) do not overlap.
Results of Z’ optimization experiments at two different temperatures are shown below. First, we set the temperature dial to 26˚C. On the left half of the 384-well plate, wildtype yeast cells grow to appreciable numbers, literally lighting up the plate.
On the right half of the plate, the PIGA mutant yeast cells barely grow, resulting in a huge delta between wildtype and mutant, a numerical value called Z prime (Z’). Adding 0.2% DMSO (dimethyl sulfoxide), the solvent in which all test compounds are solubilized, has no effect whatsoever.

This PIGA temperature-sensitive mutant is, as advertised, quite picky about the thermoset. In fact, there’s a razor-sharp gradient between life and death. The mutant’s growth defect is extremely pronounced at 26˚C as shown above. But there is no difference in growth rate between wildtype and the PIGA mutant at 28˚C, as shown below. Somewhere between 26˚C and 28˚C, the growth flip switches on.

Convinced we could maintain the temperature just right, we proceeded to run the Pharmakon screen last week. Given the distributed nature of our yeast lab operations, we had to use an incubator at SFBL instead of at UCSF, where we normally runs screens from start to finish at the Small Molecule Discovery Center (SMDC). If the temperature difference between the incubators at the two locations was even a half-degree, then we’d observe more growth from the PIGA mutant.
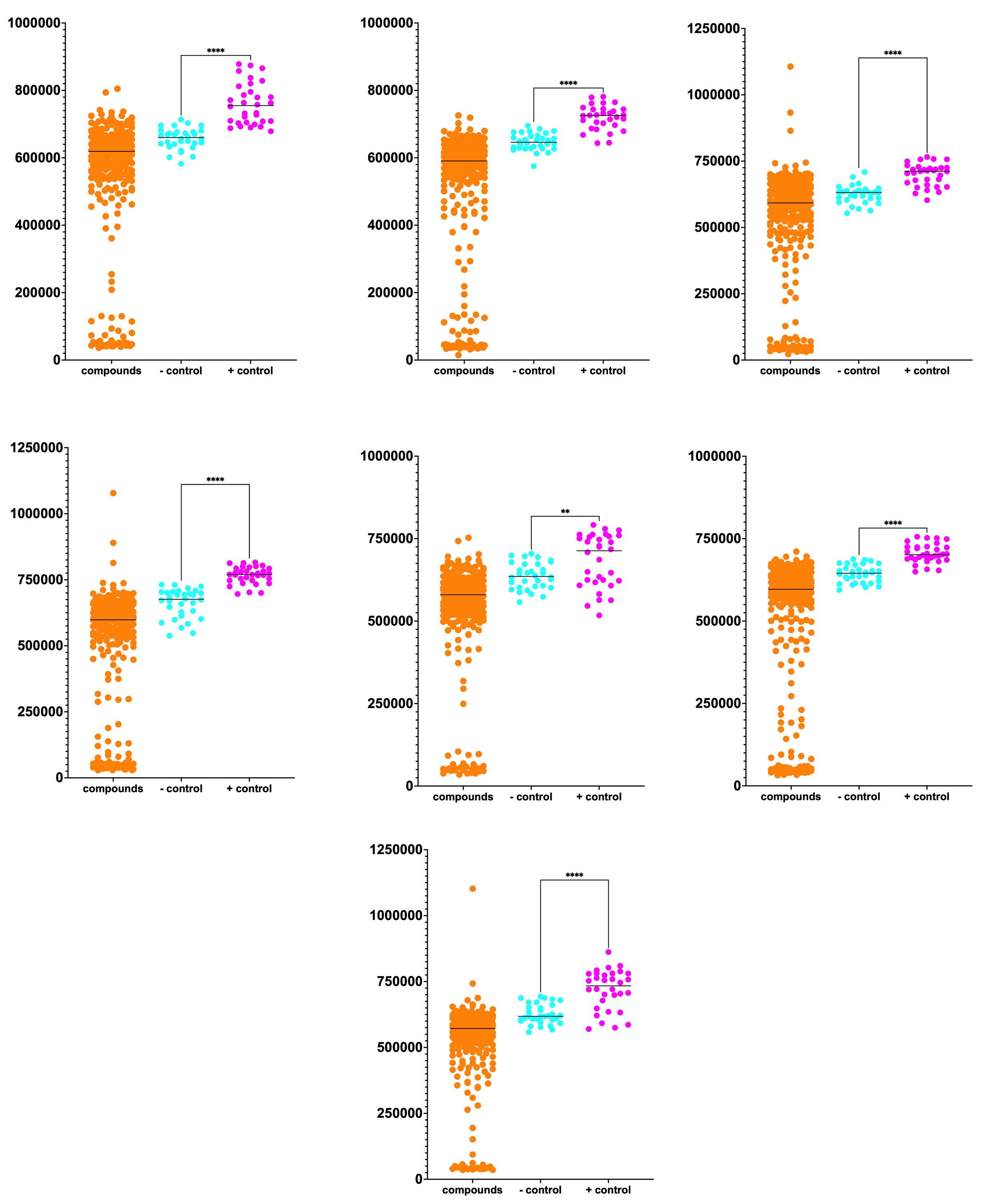
The full Pharmakon dataset of 2,250 compounds is plotted below. Unfortunately, we didn’t achieve the Z’ values that we observed in our pre-screen dry run at SMDC.

Normally we’d like to see complete separation between the negative controls (cyan circles) and the positive controls (magenta circles). Although we observe some overlap between the two distributions, the difference between the means is statistically significant.
Despite the increased growth baseline of the negative controls, nine hits highlighted in the green box rise above the pack of 2,250 test compounds. Three hits had growth that exceeded the mean growth of the positive controls by 50%. The top hit is almost twice the mean growth of the negative controls. We’ll get to the identity of these hits in a moment…
A hit rate of 9/2250 (0.4%) is somewhat lower than what we expect from previously published drug screens of yeast, worm or fly models. However — and this is quite striking — we observed 8-10X the number of sensitizers, i.e., compounds that make the PIGA mutant more sick, than expected based on all of our previous drug screens.
For example, a comparison between the PGAP3 Pharmakon dataset versus the PIGA Pharmakon dataset is plotted here.

Data from each of the seven plates comprising the Pharmakon library is plotted separately below. The sheer number of sensitizers — ~10% hit rate — suggests that PIGA may be uniquely vulnerable to the effects of chemical, and by extension genetic, modifiers.
Okay, now the part you’ve all been waiting for. Here are the 9 hits:

The top hit from the screen is deferasirox, an iron chelator (reduces iron levels). It’s used to treat iron overload caused by blood transfusions in people who have chronic anemias. We will focus on three of the hits that we believe are the most promising for immediate further study in PIGA yeast, worm and fly models, as well as in PIGA patient fibroblasts to measure ferroptosis as well as rescue of GPI protein cell surface expression:
deferasirox
undecylenic acid
naproxen
Deferasirox is an iron chelator, which means it binds to ferric iron (Fe3+). The monounsaturated undecylenic acid may also be acting as an iron binder but through a different mechanism that involves the Fenton reaction and sequestration of ferric iron in lipid aggregates called lipofuscin. Naproxen is a nonselective cyclooxygenase inhibitor aka non-steroidal anti-inflammatory drug (NSAID), which is curious because yeast don’t express COX-1 or COX-2. For that reason, let’s set naproxen aside for now.
As stated earlier, PIGA has been explicitly tied to systemic iron overload. However, the simplistic interpretation that “there’s too much iron” is unlikely to explain what is going on. Let’s quickly review the history of PIGA human genetics to understand why. In particular, two papers published within a year of each other that describe the first PIGA germline mutations in families with affected boys deserve our attention.
PIGA had been studied since the pre-genomic 1990s when it was identified as the driver gene responsible for paroxysmal nocturnal hemoglobinuria, or PNH. PNH is caused by somatic total loss-of-function mutations arising after development in blood stem cells. In humans, complete loss of PIGA in males was predicted to be embryonic lethal based on the same result in mice. Yeast lacking the PIGA gene are also inviable.
At the dawn of the clinical genomics era (2009-2013), Johnston et al (PMID 22305531) reported in 2012 the first-ever described germline PIGA mutation in a family where all three boys died as infants. They named the disease multiple congenital anomalies, hypotonia and seizures type 2, or MCAHS2.
A year later in 2013, Swoboda et al (PMID 24259288) reported another PIGA variant in an unrelated family with affected boys. However, this second family had systemic iron overload, unique skin abnormalities and none of the congenital anomalies seen in the MCAHS2 family. The authors named the new disease Ferro-Cerebro-Cutaneous disorder, which would later go on to be reclassified as PIGA-CDG.
We describe a three-generation family with three affected boys with a progressive neurodegenerative condition characterized by an infantile-onset epilepsy and encephalopathy in association with systemic iron overload.
They go on to write more about the discrepancies and commonalities between the two PIGA “founder” families in the conclusion of their paper. I bolded the most relevant sentence.
None of the reports of patients with germline mutations in genes coding for the PIG family enzymes have manifested systemic iron overload; however, patients with mutations in PIGL are reported to manifest an icthyosiform dermatitis. Since PIGA is the first enzyme in the GPI anchor biosynthesis pathway, iron overload may be specific to selective PIGA gene mutations. The family reported by Johnston et al. did not undergo iron studies (L. Biesecker, personal communication), and our data do not provide insights to the kinetics of iron load in the patients reported here. It is possible, given the similarities of neuroimaging features between the patients reported here and those reported by Johnston et al., [2012] that these represent phenotypic spectrums of the same disorder.

The most tantalizing result is that the iron chelator deferoxamine is a top hit in the PGAP3 Pharmakon screen. Deferoxamine was not a hit in the PIGA screen. In fact, deferoxamine is one of the PIGA sensitizers!
Deferasirox was neutral in the PGAP3 screen, neither rescuing nor sensitizing. As shown below, not all iron chelators are equal in terms of the physicochemical, pharmakinetic and pharmadynamic properties. Not to mention that each compound may have weaker chelation preferences for other metals like zinc and copper.

Is too much iron the problem? Or is the problem a combination of too much iron being in the wrong place at the wrong time and not enough iron being in the right place at the right time?
Earlier this week, Perlara Cure Guides Uche Medoh, Dr Kristin Kantautas and I presented all the data we have to Mariana and Paul on behalf of their son Romeo and the other PIGA-CDG families who are rallying behind this project. We learned that Romeo actually takes iron supplements, along with folic acid, and that he does well on this cocktail. Therefore, a simple linear model where PIGA deficiency leads to iron overload and toxicity is not consistent with the evidence. Something more complicated is going on.
Lysosomal storage diseases may be instructive. Sometimes a simple substrate accumulation model is adequate to explain the pathophysiology. However, there are usually second and third order effects stemming from the loss of even one critical metabolic substrate or product. There are also complex feedback (and feedforward) loops in metabolic pathways like GPI anchor biosynthesis. Hence why we’re treading carefully and thoughtfully.

Looking at the figure above, the puzzle pieces are coming into place, one gene at a time. We look forward to sharing this post with researchers and PIGA-CDG families to start to understand if we can add more nuance to “systemic iron overload” in the living laboratories of PIGA-CDG medical pioneers.
PIGS, PIGN, PIGW are next on deck for drug screens.
This is such an incredible news!
Thank you very much for the hard job and sharing!!!