FARS2 Cure Odyssey
Wendy and David launched a yeast-powered drug repurposing project for their son Alexander, who has an ultra-rare mitochondrial disease. One of the top hits is on the WHO's List of Essential Medicines.



In collaboration with


 / Twitter")

Disclaimer
The results of the FARS2 drug repurposing project that we are sharing in the spirit of open science below are novel preclinical research findings and therefore they do not constitute the practice of medicine. Please consult your physician or clinical care team if you’re considering off-label use of any approved drug. The same caution applies to nutraceuticals, supplements and “generally recognized as safe” compounds.
It took the discerning eye of a grandmother to notice that something was not quite right with Alexander, when at an early age, he had an unusual way of standing in his crib. Rather than standing on his soles, Alexander stood on the top of his feet. And as he grew, his walking led to tripping and skipping. As so often happens with ultra-rare diseases, it wasn’t until some years later that the family received a diagnosis for then 9-year old Alexander. Genetic analysis revealed that Alexander has a rare autosomal recessive disorder, FARS2 deficiency, a defect affecting mitochondrial function.
Dozen of disorders exist that impair mitochondrial function, which can result in various maladies from vision or hearing loss to muscular and cognitive deficiencies. FARS2 deficiency is one of a subset of mitochondrial disorders involving nuclear-coded mitochondrial tRNA synthetase proteins (mt-aaRS). These proteins play an essential role in protein synthesis, as they catalyze the attachment via aminoacylation of amino acids onto their cognate tRNAs.
This is a schematic of an aminoacyl-tRNA with a yeast tRNAPhe and generic amino acid. An arrow indicates where an ester bond is formed between a 3' terminal adenosine on the tRNA and the amino acid carboxyl group.

First described in 2012, FARS2 deficiency is among rarest of mt-aaRS deficiency disorders; as of this writing, only 41 variants/cases of the FARS2 gene, which codes for the phenylalanine tRNA synthetase gene, have been reported, putting it in the ultra-rare category. Mt-aaRS variants all do pretty much the same thing—disrupt the proper translation of proteins in the mitochondria, which in turn affects energy production in the form of ATP in cells throughout the body. However, the brain and the musculature appear to be the greediest organ systems, as these disorders most often result progressive motor and cognitive disabilities. Two forms of FARS2 disorders have been described: early onset epileptic mitochondrial encephalopathy, an aggressive form, appearing in the very young, and a late form characterized by spastic paraplegia. The early onset form is more prevalent—occurring in about two thirds of the cases — and has a poorer prognosis, often accompanied with developmental delays and various brain abnormalities.
Mt-aaRs disorders are inherited in an autosomal recessive fashion. Most patients are compound heterozygotes, which simply means they have different mutations in the two copies of the FARS2 gene, one originating from each parent. Although the actual amino acid changes caused by the mutations are known, the disease mechanism is not well understood. For one thing, it’s unclear why certain variants give rise to the more aggressive form.
By performing trio exome analysis of Alexander’s family (mother, father, child), researchers discovered two unique mutations in Alexander’s FARS2 genes; a paternally inherited nonsense mutation (Q216X) and a maternally inherited missense mutation (P136H). The Q216X gives rise to a truncated protein near the midpoint of the protein, thus removing both the catalytic site and anti-codon binding domain while the P136H variant results in a nonconservative amino acid replacement in what is otherwise a conserved region of the protein—- which suggests functionality might be affected, since Nature chose to conserve that part of the protein.
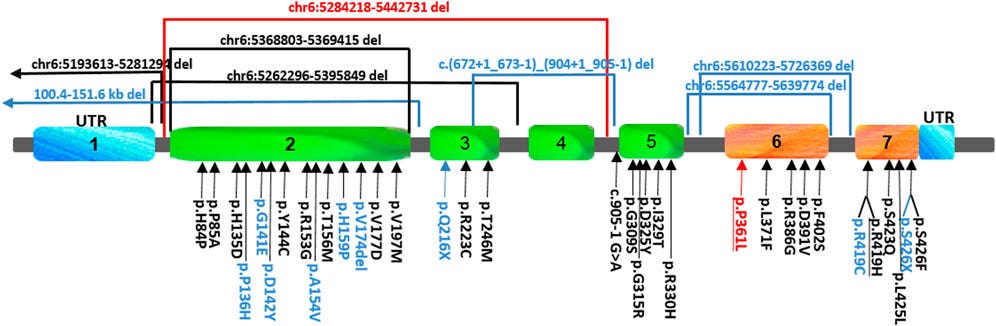
This schematic view of the location of the FARS2 variants reported to date shows that they are scattered rather uniformly throughout the protein. Those found either in epileptic or spastic patients are underlined, and those found only in epileptic patients are indicated in black, while the variants associated only with spastic paraplegia are indicated in blue.
As FARS2 syndrome goes, Alexander had what would be considered a mild case, lacking cognitive decline or debilitating epileptic encephalopathy, seen in the more severe cases. Looking for some explanation for this, the Cedars-Sinai group expressed the proteins and measured their activity in vitro, and found some residual activity with the P136H protein. (The Q216X proved impossible to purify, but given that it lacks two essential parts of the protein, it would be surprising if it were to have any activity).
And now for some good news. As a group, these proteins are conserved evolutionarily, which means that it might be possible to crack the code on the connection between genotype and phenotype is some simple model systems. In Drosophila, for example, Chinese researchers found that knocking out FARS2 two entirely led to developmental delays and seizures. Drilling down a little more precisely, they showed that one human variant linked to early onset epilepsy in people (pG309S) when expressed in flies, led to seizures, while another associated with spastic paraplegia (pD142Y) affected locomotion.
FARS2 is also conserved in Saccharomyces cerevisiae, aka our old friend budding yeast. The yeast version of FARS2 is called MFS1. Dusting off our established mitochondrial gene knockout yeast avatar playbook — see SURF1 and AFG3L2 examples — we could hit the ground running using off-the-shelf MFS1-knockout yeast mutants.
We performed a 96-well-plate growth assessment in 2% lactate media of an allelic series at two temperatures (30˚C vs 37˚C) and two timepoints (24 h vs 48 h). The allelic series consists of a wildtype homozygous diploid control (MSF1/MSF1), a heterozygous knockout diploid mutant (MSF1/∆msf1), and a homozygous knockout diploid mutant (∆msf1/∆msf1). As expected, the homozygous diploid knockout mutant exhibited a severe growth defect under all conditions tested.

We advanced to the next gate: a seeding density experiment in 384-well plates with the homozygous diploid knockout mutant at a 24-hour timepoint. Again, the results were as expected, with a large delta between the wildtype strain (black circles) and the FARS2 patient avatar (magenta squares).

The last step before progressing to the 8,400-compound TargetMol drug repurposing library is a Z’ optimization experiment that simulates the conditions of the screen. Think of it as the final dress rehearsal before opening night.
Everyone hit their mark, as demonstrated by the stark night-and-day absolute difference between the growth of the wildtype strain and the growth of the FARS2 patient avatar. Notably, the well-to-well variance is low, resulting in Z’ values close to the gold standard value 0.5.
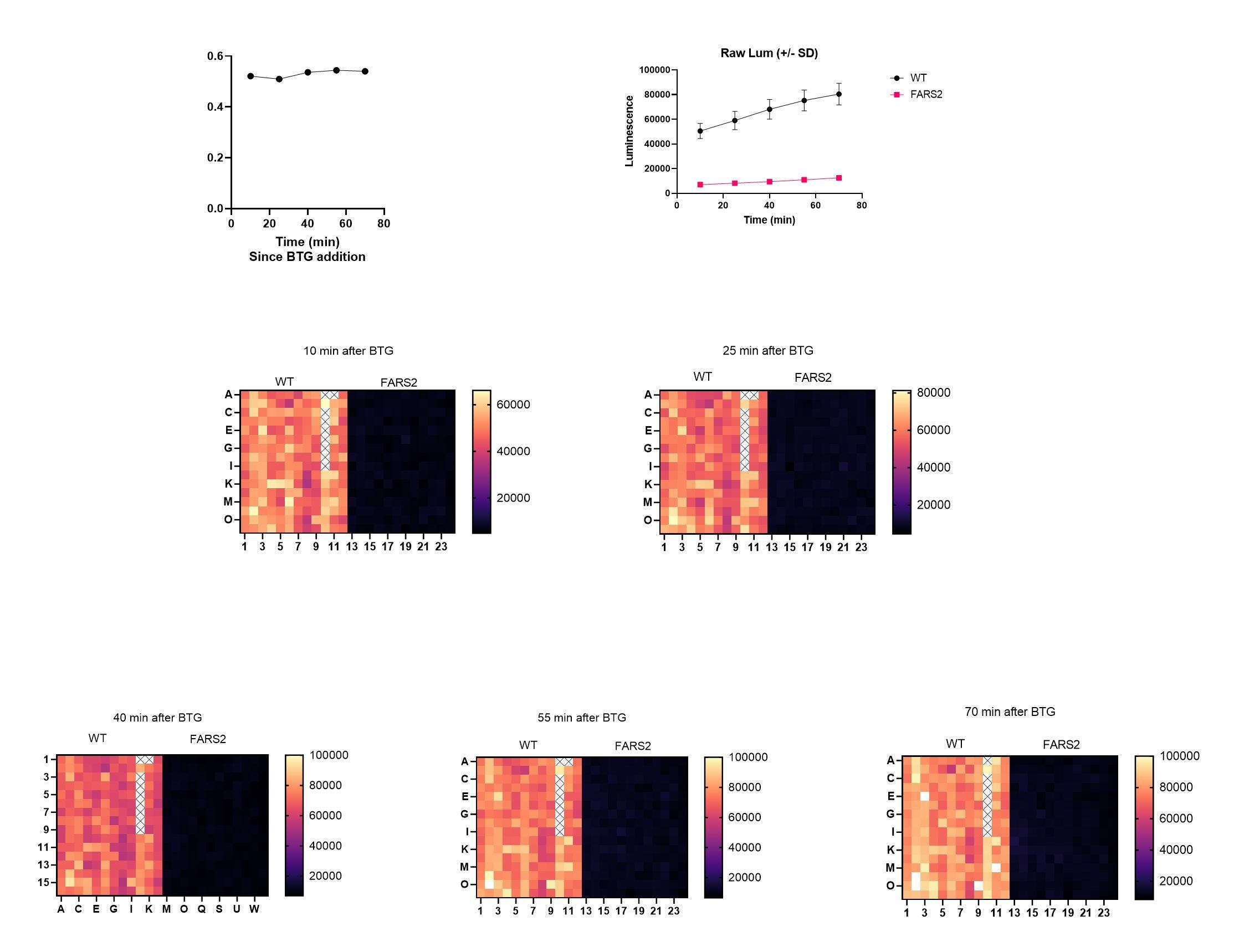
Below is a summary plot of the 8,400-compound TargetMol screen. As per usual, the orange circles represent individual test compounds and the FARS2 knockout yeast avatar. The negative control is the FARS2 knockout yeast avatar treated with DMSO (cyan circles), the solvent in which all test compounds are dissolved. The positive control is the wildtype yeast treated with DMSO (magenta circles). While there was some slippage of wildtype yeast that grew sub-optimally, the FARS2 knockout yeast avatar grew consistently poorly, as expected.

The identities of the rescuers are listed below. We created two buckets: strong-to-moderate rescuers (boxed in dark blue) and moderate-to-weak rescuers (boxed in light blue). Several mechanistically and structurally related classes of rescuers are evident even to the chemically untrained eye. They include three types of fatty acids, and a sprawling family of plant-based Chinese Traditional Medicines called steroidal saponins.

Let’s focus our attention on the top two hits that have the largest effect size: perifosine and miltefosine.
Both compounds belong to a pharmacological class called lysophosphatidylcholines, or LPCs. Technically, they are classified as alkylphosholipids. LPCs are a modified form of the naturally occurring phospholipid called phosphatidylcholine (PC) where one of its two fatty acids tails is lopped off. LPCs mimic PC inside the cells, inhibiting the enzymes that produce PC and gumming up the protein transporters that normally control the flow of lipid tides from one organelle to another.
Since 2014, miltefosine (trade name Imavido) is a FDA approved oral drug for use as a 28-day treatment regiment for leishmaniasis, a parasitic infection. It was originally developed in the 1970s and 1980s as an anti-cancer agent. It was approved in India as an antileishmaniasis therapy in 2002. Miltefosine sits on the WHO’s List of Essential Medicines. For the avoidance of doubt, it is not FDA approved for chronic use in any disease indication.

Perofosine and miltefosine are not the only lysophosphatidylcholines that scored as FARS2 rescuers in the yeast screen. All of the compounds whose structures are displayed below are united by the presence of a positively charged choline or terminal quaternary amine, but there is tolerance for a range of acyl chain lengths. What’s more, the phosphate group is dispensable for activity. Interestingly, two versions of phosphatidylcholine are also rescuers, albeit weak ones. The unifying structure-activity relationship appears to be phosphatidylcholine mimicry.
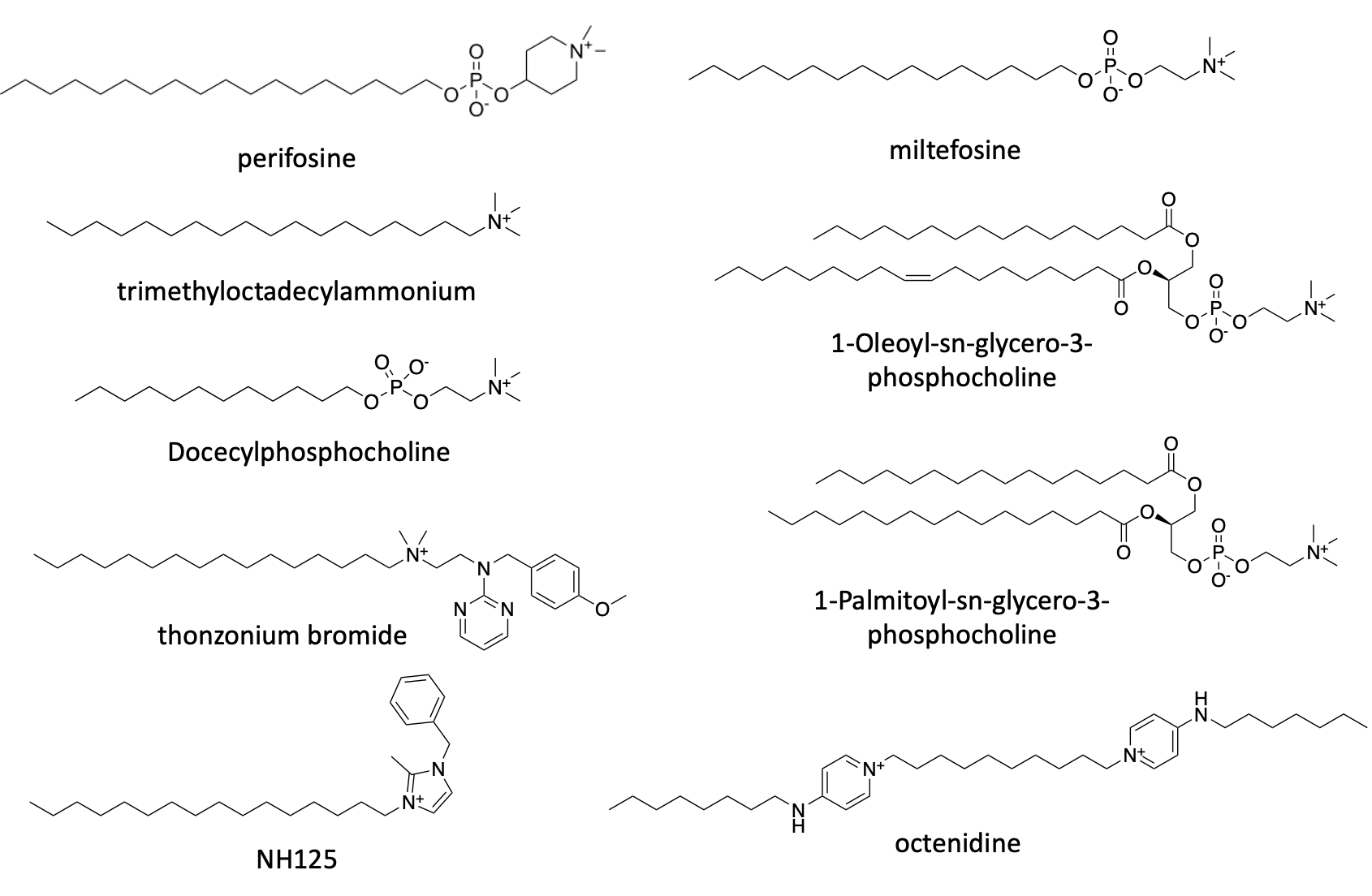
The screen also yielded a pair of sphingolipid analogs, amiselimod and fingolimod. Phospholipids and sphingolipids both have nitrogen-containing head groups like choline or ethanolamine. Why are positively charged (cationic) lipids protective in FARS2-deficient yeast? We’ll return to this question momentarily.

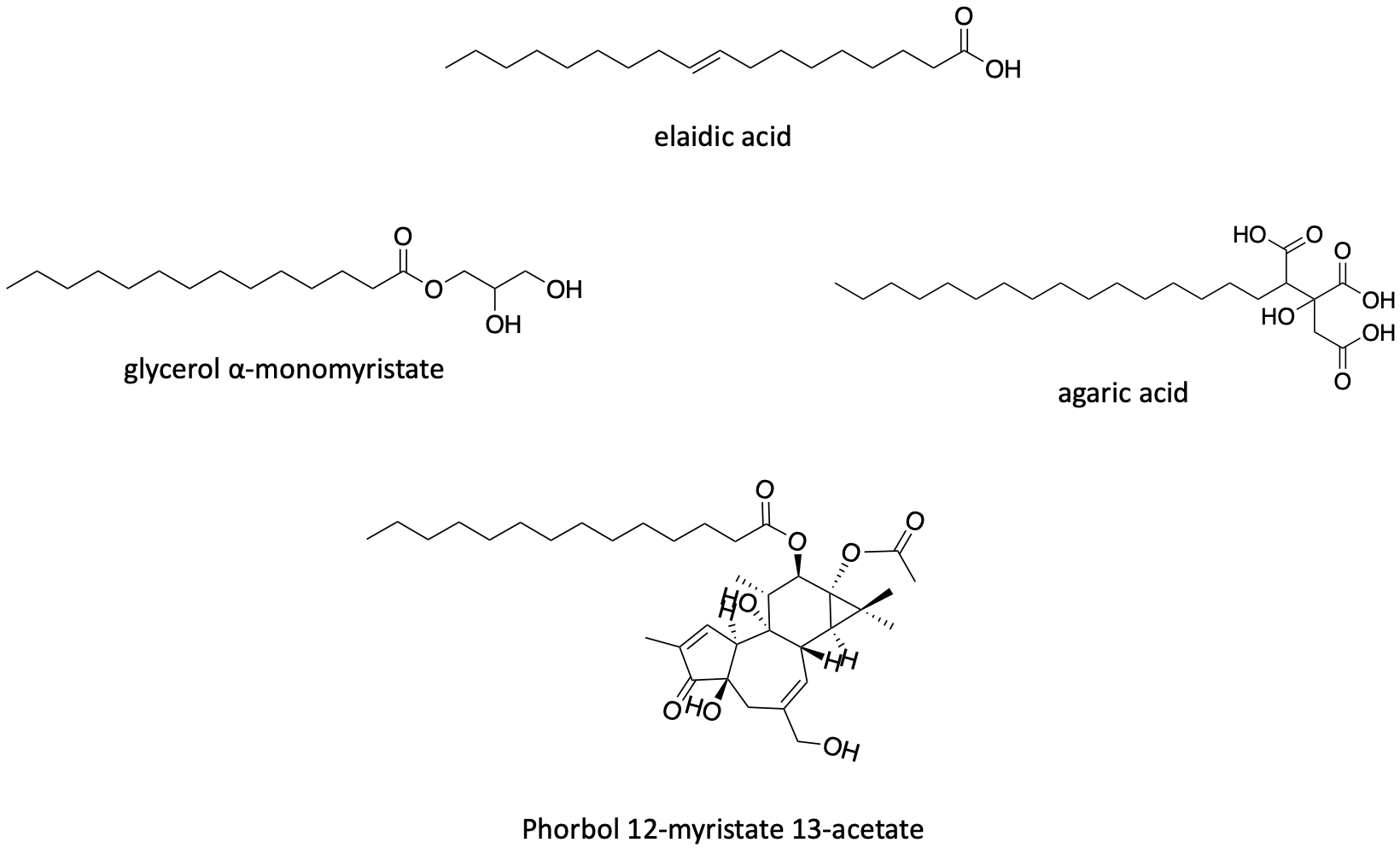
Curiously, in addition to the cationic fatty acids shown above, the FARS2 yeast screen also yielded a group of structurally related negatively charged (anionic) fatty acids. Elaidic is the trans isomer of oleic acid, which is famously found in olive oil. Glycerol alpha-monomyristate is a commonly used food additive. Agaric acid is a natural product found in fungi but is not generally recognized as safe (GRAS). Phorbol 12-myristate 13-acetate (PMA) is a laboratory tool compound historically used to stimulate the protein kinase C (PKC) pathway. Agaric acid and PMA shared a diacylglycerol backbone.
Finally, there are two groups of steroidal saponins, which are Chinese Traditional Medicines derived from plants. The first group shares an oleanane scaffold. Intriguingly, the aglycosidic Hederagenenin appears alongside its monosaccharide (Calenduloside E), disaccharide (Momordin Ic and Ginsenoside Z-R1) and trisaccharide (Hederacolchiside A1 and Raddeanin A) versions. There doesn’t seem to be a preference for the number of sugars attached to the steroidal core.

The second group of saponins belong to the sarsasapogenin family. As is the case for the first group of saponins, 1-3 sugar attachments are seemingly equally tolerated: the monosaccharide (Timosaponin AI), disaccharide (Timosaponin AIII) and trisaccharide (Dioscin and Pennogenin 3-O-beta-chacotrioside) versions all rescue.

At this juncture, we can only speculate about mechanisms of action. It is a noteworthy coincidence that 5 out of the top 10 rescuers are choline-bearing lipids, and 3 out of the top 12 rescuers are steroidal saponins. A 2010 paper in on the effects of miltefosine (and other LPCs) and lipid metabolism in a well studied hepatic carcinoma cell line (HepG2) found that miltefosine changes the balance between choline-bearing lipids and cholesterol. Specifically, miltefosine decreases the levels of phosphatidylcholine and sphingomyelin, which also has choline head group, while at the same time increasing the levels of cholesterol.
Why might shifting the balance of lipids matter to the mitochondria? Membrane lipids with positively charged head groups interact with other membrane lipids with negatively charged head groups. More cholesterol in mitochondrial membranes might somehow increase the cell’s capacity to deal with misfolded mitochondrial membrane proteins that build up when there’s not enough tRNA-Phe to fuel protein production at full speed and high fidelity.
Next steps on the preclinical research front: test the top 10-15 yeast hits in Alexander’s fibroblasts (and potentially myoblasts) using the Seahorse instrument followed by mitochondrial lipidomics and mitochondrial protein stability studies.
Of course, for the families, treatment for FARS2 can’t come soon enough. Although Alexander has not experienced some of the more serious symptoms — with the infantile form, children die before the age of two — his disease is not without consequence. Now 16, Alexander lives normally, his mother describes him running up and down subway steps on a recent trip to Manhattan. Yet, she also reports that is walking has worsened lately — he is more apt to lose his balance and he is finding it hard to lift his legs or step sideways. And people are noticing in a new way. Before he had a little bit of a weird walk and people might or might not notice. But now people are seeing him as disabled, according to his mother — offering golf carts, pointing out elevators, something that has not happened to Alexander so far, who has refused to identify as handicapped. His family as well has not applied for services or handicapped parking. Whether access to intervention with these therapies will come in time to allow him to retain his identity — “that’s our hope.”
Absolutely incredible to read this article, as it gives me hopes for my 15 month old son who has just been diagnosed with epilepsy, with rare gene affected FARS2. Neurologist have a hard time controlling seizures and told me there is no cure for the illness that is unknown causing this. He has been in the hospital since 30 may 2024 as I noticed a bit of a tremor in his right arm and hand, and has now progressed as he is having full tonic clinic seizures, neurologist have tried many anti epileptic meds and nothing is working, I read this article and this brought hope. I don’t have much financially, but have hope for cure and treatment.
Mother of a lovely boy who just wants to thrive and live life to the fullest.
Nuraawale@gmail.com