Fuel It Still
Boosting levels of the metabolic fuel NAD+ rescues DHDDS deficiency in a yeast avatar. 3 pioneer N-of-1 studies are now underway. Rosie's N-of-1 is yielding results after 12 weeks of NMN treatment.


In collaboration with



 / Twitter")

Disclaimer
The results of the DHDDS drug repurposing project that we are sharing in the spirit of open science below are novel preclinical research findings and therefore they do not constitute the practice of medicine. Please consult a physician or clinical care team if considering off-label use of any approved drug or compassionate use of any experimental drug. The same caution applies to nutraceuticals, supplements and “generally recognized as safe” compounds.
Just one spark from a bolt of genetic lightning can ignite an inherited metabolic disease that wreaks havoc across metabolism. Down to the unit of the cell and bubbling up as symptoms at the level of tissues and organs. Take the case of the ultra-rare genetic disease DHDDS deficiency.
The DHDDS gene gets its acronym from the enzyme it encodes: dehydrodolichyl diphosphate synthase. It’s responsible for the conversion of the one-to-many metabolite farnesyl to polyprenol, which is the precursor of dolichol. Dolichol serves as the foundation on top of which a glycan chain is elaborated by an assembly line of worker bee enzymes, one sugar brick at a time, as summarized in the diagram below.

No dolichol, no glycoproteins, yes disease. But how exactly?
In the simplest cases of inherited metabolic diseases (IMDs) where just one self-contained subnetwork of the grid is knocked out by mutation, dietary supplementation or restriction of a single metabolite can be curative if implemented in infancy. Think biotin supplementation for biotinidase deficiency, and phenylalanine restriction for phenylketonuria (PKU). Rephrased in the parlance of biochemists, dietary supplementation becomes substrate replacement therapy, and dietary restriction transforms to substrate reduction therapy.
Longtime readers will remember that substrate reduction therapy constitutes the therapeutic basis for treating SRD5A3-CDG with atorvastatin. SRD5A3 happens to be the enzyme two stops down the line from DHDDS. We hypothesize that lowering the flux of the upstream metabolite mevalonate prevents toxic overproduction of polyprenol, which competes head-to-head with dolichol leading to immature glycan formation and hypo-glycosylation. Polyprenol is one of many downstream products generated from mevalonate, whose biosynthesis by the enzyme HMG-CoA is inhibited by statins.
However, the critical difference between SRD5A3 and DHDDS is that human life is incompatible with complete loss of DHDDS, while SRD5A3 has a close cousin called SRD5A2 that partially backfills the loss of SRD5A3. In other words, there must always be some residual DHDDS enzymatic activity in every cell, even if only a modicum of normal levels. Instead of slowing down the tempo, what if the remedy in cases like DHDDS deficiency is to make the enzymes positioned after DHDDS in the assembly line work harder, i.e., substrate enhancement therapy?
A single hobbled enzyme like DHDDS can be a flashpoint for multidirectional disruptions that sweep unevenly across the grid of cellular metabolism, surging flows at some chokepoints while tripping circuit breakers at others; simultaneously causing there to be too little or too much of key metabolites; and, therefore, demanding multi-pronged therapeutic strategies that restore balance to the diseased metabolic network as a whole.
For most IMDs, including DHDDS, the toxic presence of unwanted and normally transient metabolites must certainly be factored into the pathophysiology equation, but so too must the toxic absence of essential metabolites. Especially central hub metabolites that are rate-limiting precursors for numerous downstream metabolic pathways. In the case of DHDDS, that means coping with rock-bottom levels of dolichol.
The simplicity and beauty of unbiased drug repurposing screens of yeast patient avatars mean that we don’t need to know a priori which enzymes act as the key circuit breakers or backup generators when a given node in metabolism is compromised. We don’t need to know a priori whether we need to increase metabolic flux or decrease metabolic flux at those critical nodes in the grid. Fortunately, yeast are here to give us answers.
AI certainly couldn’t know the answer because there is no training data on under-studied or new-to-medicine ultra-rare diseases, a state of affairs describing the majority of the 1,500 inherited metabolic diseases. Per Sam Altman’s recent AGI dawn forecast, for at least the next 1,000 days AI has to play nicely with OI — Organic Intelligence, which is a cute way of saying we need to learn all we can from predictive disease models like yeast, worms and flies. Gradually, reinforcement learning with patient avatar feedback will turn shaky in silico hallucinations into undeniable in vivo validations. But we have to create the knowledge base first!
Cure DHDDS is an Anglo-American alliance between the Dixon family in the UK and the Manville-Gourley family in US. The latter happen to be the founding members of the band Portugal The Man. Proving once again that genetic lightning can strike anyone. Anytime. Anyplace.
When it does it can reveal a hidden superpower in parents who propel themselves into the multi-faceted role of caretaker, advocate, fundraiser, scientist, and, in the fullness of time, biotech founder and CEO. No one else will care as much about a rare disease than those who are personally affected by it. Instead of running away from this hard truth, let’s embrace it and unleash the uncapped potential of patients as the platform.
Over the past two years, Perlara and pioneer IMD families have ironed out a three-phase workflow for yeast-powered drug repurposing screens where hits from the screen can be rapidly translated to kiddos based on the weight of the evidence from a yeast screen combined with a first-principles approach to the practice of medicine. Let’s not forget that we also need objective, quantitative, and data-rich functional outcome measures to replace outdated, pen-and-paper, one-shot assessments like the ataxia rating scale ICARS. I’ll return to this key point at the end of this post.
In the first phase of drug repurposing workflow, growth defects of yeast avatars are verified and shown to be statistically separable from wildtype yeast. In the case of DHDDS, we were fortunate to be able to leverage previously published and experimentally vetted DHDDS yeast avatars.
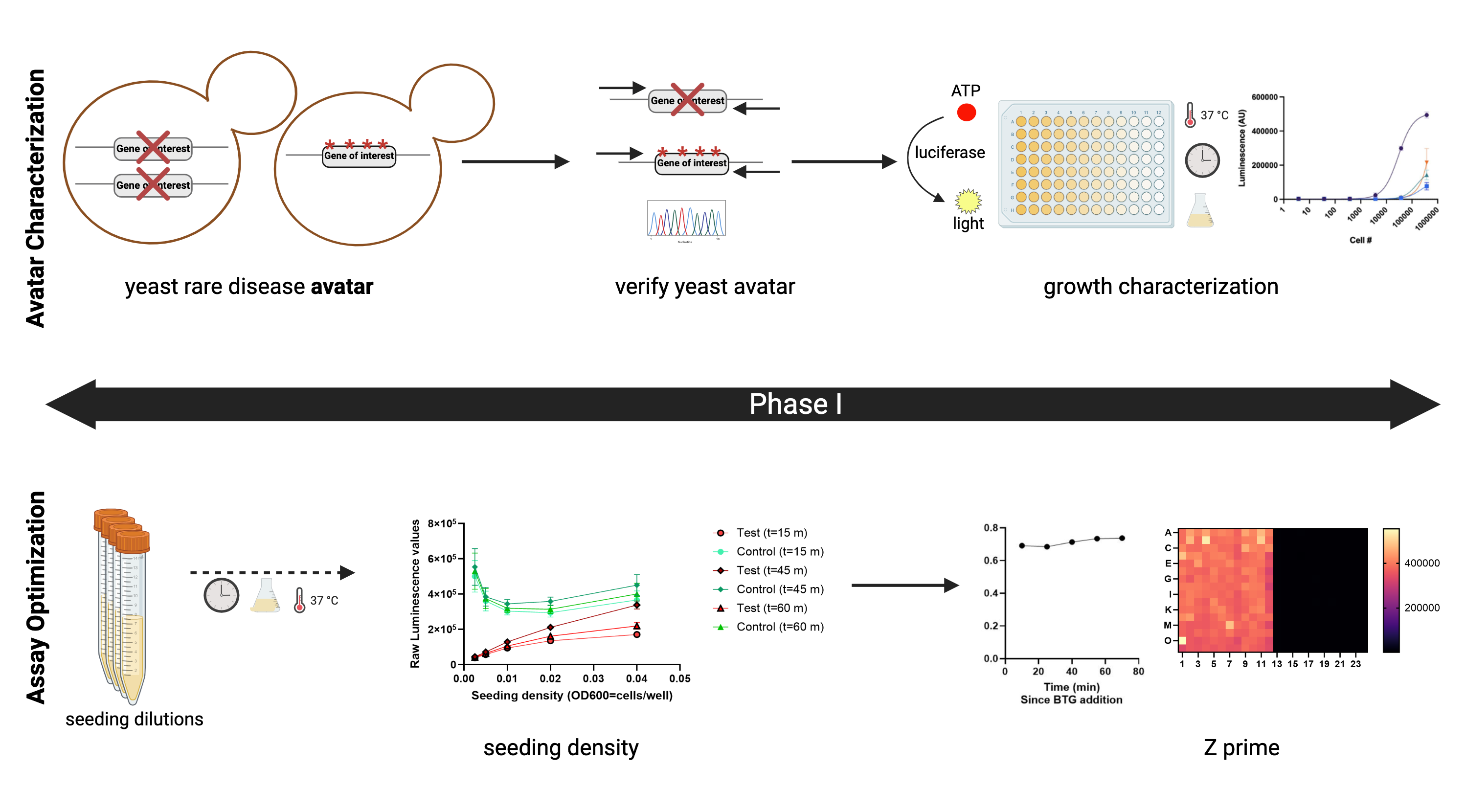
In the second phase, we screen the 8,400-compound TargetMol library. It has served us and families well. In all eight yeast-powered drug repurposing screens we’ve performed to date, we’ve found at least one clinically actionable hit, almost always an over-the-counter nutraceutical/supplement, starting back in 2018-2019 with PMM2-CDG and the discovery of alpha-hydroxy-cinnamic acid (aka cinnamon), which in turn led to epalrestat.
Examples of parent-led studies we’ve disclosed so far on our Substack include antioxidants for PGAP3, steroidal saponins for AFG3L2, disulfiram for ADSL, and ascorbyl palmitate for PIGS-CDG. For PIGN-CDG, PIGW-CDG, and ALG11-CDG, we have active 1-to-N studies underway with compounds whose identities we will disclose in due time.

In the third phase, we generate a hit list and carry out hit validation experiments such as dose responses, and if necessary to build conviction — as proved to be the case for DHDDS — mechanistic studies that include evaluating a therapeutic hypothesis like substrate enhancement.

It took 12 months to go from project launch to the first N-of-1 parent-led observational study that began over the summer. Some projects move faster and multiple steps can be skipped, for example hit validation and mechanistic studies. We took more time because we made a novel discovery along the way, as I’ll describe. The DHDDS drug repurposing project timeline is shown below.

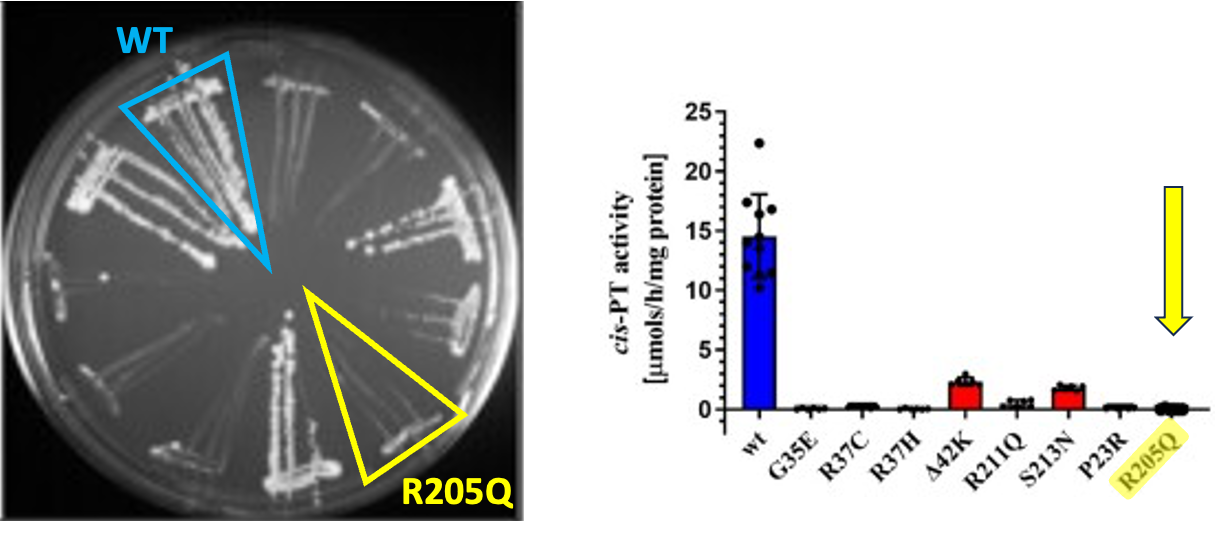
We had a little help from some friends in the Grabinska lab at Yale. They created the yeast DHDDS avatars in the first place. I will focus on one DHDDS variant, R205Q. As you can see in the original publication from 2022, the R205Q avatar is extremely sick — it barely grows — and its DHDDS protein barely has detectable enzymatic activity (referred to as cis-PT). We advanced the DHDDS R205Q yeast avatar to a drug repurposing screen. The severe presentation of R205Q creates a reproducible night-and-day growth difference with the wildtype.
We previously shared high-level results just under a year ago.
The plot below shows the distributions of growth values from the DHDDS TargetMol screen. The positive control (aka wildtype) are the magenta points, soaring above the rest. The negative control (aka R205Q avatar plus placebo) are the cyan points way down below. Each of the orange points is one of the 8,400 tested compounds.

I highlight in yellow two metabolic cofactors, NADH and NADPH, as being in the top 10 rescuers of the DHDDS R205Q avatar. Notably, these cofactors are required for dolichol biosynthesis. In hit validation dose-response experiments, NADPH repeated but NADH did not. (Solubility and stability issues in yeast media are the likely culprits).

NADH and NADPH have never come up as a pair of rescuers in any other TargetMol screen we’ve done. We have observed UTP and CTP as rescuers in a few other CDG screens, but never alongside NADH or NADPH. When looking more closely at the dolichol production pathway and the list of top rescuer compounds, we were struck by the enrichment for enzyme cofactors that stimulate activity of the enzymes that are immediately downstream of DHDDS.

It suggested to us that the scarcity of polyprenol forced the enzymes downstream of DHDDS to work extra hard to utilize whatever polyprenol eekes out of the hobbled DHDDS protein. CTP just so happens to be a cofactor for dolichol kinase, the enzyme one step after SRD5A3 that phosphorylates dolichol and primes it to receive sugar building blocks. In the dose-response experiment above, we see impressive rescue of the DHDDS R205Q yeast avatar by CTP. (The high luminescence values suggests that some of the rescue effect is an artifact).
It had been thought that the SRD5A3 enzyme converts polyprenol directly to dolichol. But a recent paper smashed that model to bits, assigning the conversion of polyprenal to dolichal to SRD5A3, and implicating another enzyme called DHRSX as the missing link that bridges polyprenol to dolichol via a pair of side reactions that involve polyprenal and dolichal intermediates.

Remarkably consistent with our DHDDS TargetMol hit list, the newly recognized enzyme DHRSX utilizes both NAD+ and NADPH as cofactors. The former in the reaction that converts polyprenol to polyprenal, and the latter in the reaction that converts dolichal to dolichol. In fact, DHRSX works upstream and downstream of SRD5A3, which itself is a NADPH-dependent enzyme.

Altogether, these data lead us to hypothesize that steady-state NAD+ levels in DHDDS-deficient cells are not sufficient to drive conversion of polyprenol to dolichol. Therefore, the cell needs to boost NAD+ levels in order to drive flux through the dolichol production pathway. This ensures that every last molecule of polyprenol is immediately sopped up by the DHRSX and SRD5A3 tandem bucket brigade, which have to work overtime to compensate for diminished DHDDS activity.

Much to our delight, we observed robust growth rescue of the DHDDS R205Q yeast avatar by NMN, niacin and NMMH (in that order). NR and NAM didn’t rescue but that could be a bioavailability issue in yeast, or yeast vs human NAD+ biosynthesis and salvage pathway evolutionary divergence. The NAD+ precursors we tested in yeast are all available as over-the-counter supplements. NMN is particular is being pursued for multiple disease indications, and is often bound up in conversations (and claims) about healthspan extension and longevity.

The proof is in the pudding, or in the iPhone as it were. A major learning from our first N-of-1 studies is that the index (pioneer) patient, i.e., the first N-of-1, may not be representative of the median patient with the same disease. In fact we should even expect an “over-response” that may be amplified by environmental factors like an optimized diet, access to supplemental physical and occupational therapies, and a family’s level of hope and optimism for the future.
We believe the way to mitigate over-indexing on the outcome of a single N-of-1 is to start multiple N-of-1’s in parallel. If the same improvements are observed in three kiddos, it becomes harder to explain away the results as a fluke or an exaggeration of the true responder rate. So Mel’s two children, Rosie and Tom, and Zoe’s daughter Frances, are the three DHDDS NMN pioneers. Over the summer, Rosie’s NMN N-of-1 started first, followed by Frances’s NMN N-of-1 a few weeks later. Rosie’s brother Tom started his NMN N-of-1 several weeks ago.
Working with Dr Eva Morava, the pioneer DHDDS families are exploring the sensitivity and robustness of at-home video capture as a universal functional outcome measure that is completely disease-agnostic. One of the assessments is the “walk the line” test.
Here’s Rosie’s baseline video before she started taking NMN:
And here’s Rosie after 11 weeks on NMN. Do you notice a difference?
In two weeks, the DHDDS NMN pioneers have an appointment with Dr Morava at Mt Sinai in New York City to review the totality of the data and make a concrete plan for next steps.
Let’s conclude by taking a moment to acknowledge the predictive power of yeast avatars and the ocean-parting powers of pioneer families battling a rare disease. That potent combo allowed us to springboard rapidly and responsibly from a compound hit list to a data-driven intervention in the real world. Onward!